




la Newsletter

Annalia Cianflone1, Daniela Braconi2, Annalisa Santucci2, Rossella Parini3,4
1S.S. Citogenetica e Genetica Medica, ASST-Monza, Ospedale San Gerardo; 2Dipartimento di Biotecnologie, Chimica e Farmacia, Università degli Studi di Siena; 3Ambulatorio Malattie Rare dell’adulto, ASST-Monza, Ospedale San Gerardo; 4IRCCS San Raffaele Istituto Telethon per la terapia genica (SRTIGET), Milano
Alcaptonuria: focus su una malattia genetica ultra-rara | Aciduria...
Aciduria omogentisica, ocronosi e osteoartropatia costituiscono la...
 L’alcaptonuria (AKU, OMIM 203500) è una malattia genetica ultra-rara a trasmissione autosomico-recessiva causata dal deficit dell’enzima omogentisato 1,2-diossigenasi (HGD, EC 1.13.11.5) che trasforma l’acido omogentisico (HGA) in maleilacetoacetato (1) (Fig. 1).
L’alcaptonuria (AKU, OMIM 203500) è una malattia genetica ultra-rara a trasmissione autosomico-recessiva causata dal deficit dell’enzima omogentisato 1,2-diossigenasi (HGD, EC 1.13.11.5) che trasforma l’acido omogentisico (HGA) in maleilacetoacetato (1) (Fig. 1).
L’enzima è espresso prevalentemente a livello epatico e renale, ma anche in prostata, piccolo intestino, colon, cellule osteoarticolari e cervello (2) e fa parte della via di degradazione di fenilalanina e tirosina. Il suo deficit causa un aumento dei livelli di HGA e la deposizione di un pigmento scuro (rosso-blu-marrone) prevalentemente a carico dei tessuti connettivi, fenomeno noto come “ocronosi” (3) a cui si associano una varietà di manifestazioni cliniche quali: pigmentazione scura della sclera e del padiglione auricolare, artrosi precoce, valvulopatia, calcificazione delle coronarie, calcoli renali e talvolta insufficienza renale.
Epidemiologia
La prevalenza globale dell’AKU è di 1:250.000 - 1:1.000.000 (https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=56), e la malattia colpisce tutti i gruppi etnici. La prevalenza dell’AKU in Italia non è nota. Nella Repubblica Dominicana e in Slovacchia è circa 1:20.000, mentre negli Stati Uniti ed in Europa è sicuramente più bassa, circa 1:100.000 [1].
Genetica e correlazione genotipo-fenotipo
Più di 250 varianti patogenetiche del gene HGD sono descritte in un database dedicato (http://hgddatabase.cvtisr.sk). La maggior parte dei pazienti sono doppi eterozigoti, cioè portatori di due diverse varianti, e solo una minoranza è omozigote, cioè portatrice della stessa variante sulla coppia di geni HGD (4). Circa il 65% delle mutazioni sono missenso, determinano cioè una singola sostituzione aminoacidica (4) e si traducono nella sintesi di una proteina più o meno funzionante a seconda della struttura proteica finale. Perciò le mutazioni missenso possono essere sia lievi che gravi. Altri tipi di mutazioni del gene HGD sono nonsenso (4%), frameshift (11%), splicing (14%), e grosse delezioni (5%): la maggior parte di queste sono gravi (4). Non è ad oggi riconosciuta una correlazione genotipo/fenotipo (4), e persino i fratelli mostrano età di esordio e diversa gravità dei sintomi a fronte delle stesse mutazioni genetiche e di una dieta simile.
Fisiopatologia
A causa della ridotta attività dell’enzima HGD, in AKU si riscontrano livelli sistemici elevati di HGA. L’eccesso di HGA viene in gran parte rimosso per via urinaria (3) tuttavia, con il tempo e attraverso meccanismi non del tutto chiariti ma che verosimilmente coinvolgono fenomeni di ossidazione e aggregazione, l’HGA non escreto porta alla produzione di un pigmento simile alla melanina che si deposita sul collagene, impartendogli la tipica colorazione scura (ocronosi). La pigmentazione è visibile ad occhio nudo in alcuni distretti corporei (occhio, cute, orecchio), ma colpisce in modo significativo soprattutto le grandi articolazioni ed il sistema cardiovascolare.
Uno degli ostacoli principali alla comprensione dei meccanismi fisiopatologici dell’AKU è rappresentato, ovviamente, dalla rarità dei campioni ocronotici e dalle tecniche invasive richieste per la loro raccolta. Tali tessuti sono intrinsecamente difficili da studiare in quanto molto rigidi e fragili (quindi difficilmente manipolabili) e lo stesso pigmento è resistente alla degradazione chimica ed enzimatica (impedendone la caratterizzazione) (2). Tuttavia, chiarire i meccanismi molecolari dell'AKU e dell'ocronosi aiuterebbe a meglio comprendere la malattia e migliorare la qualità di vita dei pazienti sviluppando terapie farmacologiche mirate. In quest’ottica, l’allestimento e la caratterizzazione di vari modelli cellulari in vitro ed ex-vivo sono stati fondamentali per dimostrare che stress ossidativo, infiammazione, amiloidosi secondaria, alterazioni del citoscheletro, ciliopatia e neoangiogenesi hanno un ruolo nella fisiopatologia e nella progressione dell’AKU (2,5,6).
Storia naturale
Aciduria omogentisica, ocronosi e osteoartropatia ocronotica costituiscono la triade temporalmente correlata che caratterizza l’AKU.
L’imbrunimento delle urine dovuto all’ossidazione dell’HGA è l’unica manifestazione clinica osservabile in età pediatrica che può consentire la diagnosi entro il primo anno di età, ma questo accade solo in circa il 21% dei pazienti (1,3). Il fenomeno è osservabile sia nel pannolino che nell’urina conservata ed esposta all’aria ma, poiché lento, può passare inosservato.
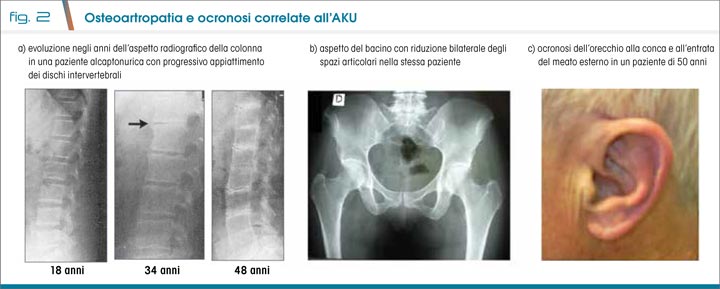
Nelle prime due decadi di vita il paziente cresce normalmente e non lamenta particolari disturbi. Nella terza - quarta decade possono presentarsi dolori alla schiena per un iniziale danno della colonna e artralgia delle grosse articolazioni. Dal punto di vista radiologico le alterazioni articolari si evidenziano dopo i 30 anni e l’età media di intervento per protesi articolare è 55 anni (3). L’artrite dell’AKU inizia alla colonna con appiattimento e calcificazione dei dischi intervertebrali e successiva fusione dei corpi vertebrali (Fig. 2a). Si osservano anche osteofiti e calcificazione dei legamenti intervertebrali. Le immagini radiografiche delle grosse articolazioni mostrano una riduzione degli spazi articolari (Fig. 2b). Sempre nella terza decade iniziano a notarsi le deposizioni di pigmento nella sclera, nella cartilagine dell’orecchio (conca e antielice) (Fig. 2c) e sulla cute delle mani in corrispondenza dei tendini sottostanti. Il pigmento è presente anche nel cerume e nel sudore (7).
Il coinvolgimento cardiaco è riconoscibile dopo i 50 anni con calcificazione o insufficienza aortica e/o mitralica e calcificazione delle arterie coronariche (8). Sono frequenti i calcoli, sia renali che prostatici, biliari e salivari. Alcuni pazienti presentano anche ripetute rotture di tendini in diversi siti, insufficienza renale e ipotiroidismo. La sopravvivenza non è ridotta rispetto alla popolazione generale ma è significativamente ridotta la qualità di vita (9).
Diagnosi
La diagnosi clinica si basa sulla presenza di urine scure, ocronosi, e artrite.
Quella biochimica, in presenza di segni e sintomi suggestivi di AKU, si basa sul riscontro di elevati livelli di HGA nelle urine (da 1 a 8 grammi/die) attraverso gas-cromatografia e/o spettrometria di massa (8). La diagnosi molecolare, necessaria soprattutto per il counselling genetico, si basa sulla identificazione delle varianti patogenetiche nel gene HGD (10).
Diagnosi differenziale
L’ocronosi dell’AKU può essere confusa con le alterazioni cutanee, acquisite e reversibili, provocate dall’uso prolungato di acido carbolico (fenolo) che si usava in passato per il trattamento di ulcere croniche cutanee. È stata descritta anche ocronosi indotta da farmaci antimalarici, antibiotici e idrochinone, agente non autorizzato in Italia, utilizzato come schiarente nelle creme di bellezza [8]. La ricerca di HGA nelle urine potrà chiarire la diagnosi.
L’artrite dell’AKU è simile alla spondilite anchilosante per quanto riguarda la localizzazione alla colonna e alle grosse articolazioni, ma l’aspetto radiologico è differente. Anche l’artrite reumatoide e l’osteoartrite hanno un quadro radiologico diverso (8).
Follow-up
Il monitoraggio clinico comprende:
- Valutazione periodica in un centro esperto di malattie rare dove alla prima visita sarà fatta una anamnesi accurata ed un esame clinico con particolare attenzione alle eventuali limitazioni di movimenti della colonna e delle grosse articolazioni.
- Visita fisiatrica per valutazione complessiva dei range of movement e dell’eventuale dolore della colonna e articolare, da ripetere una volta all’anno.
- Elettrocardiogramma ed ecocardiogramma ogni 1-2 anni se il paziente ha un’età >40 anni ed eventuale tomografia computerizzata per evidenziare possibili calcificazioni delle arterie coronariche.
- Alla prima valutazione e in caso di segni clinici, ecografia dell’addome per eventuale presenza di calcoli renali e Rx addome se sospetti calcoli prostatici.
- Una volta all’anno dosare TSH e tiroxina libera alla ricerca di un eventuale ipotiroidismo.
- Al momento della diagnosi offrire una consulenza genetica che chiarisca la natura della malattia, il meccanismo di trasmissione genetica e l’evoluzione complessiva della malattia negli anni.
La natura multisistemica dell'AKU, con problemi sia reumatologici che clinici, ha storicamente reso difficile quantificare la gravità della malattia e l'efficacia del trattamento. A tale scopo è stato sviluppato uno score clinico di severità della malattia denominato AKUSSI (Alkaptonuria Severity Score Index) e ottenuto considerando punteggi basati su caratteristiche cliniche e su specifiche evidenze della malattia a livello articolare e spinale (9). Questo score, insieme a valutazione della qualità della vita attraverso questionari standardizzati di uso comune in reumatologia (es. HAQ, KOOS, SF-36), è stato utilizzato anche per la valutazione dell’efficacia di nitisinone (9).
Terapia
Fino ad oggi, la terapia dell’AKU è stata in primo luogo sintomatica: trattamento personalizzato dei dolori articolari, fisioterapia per mantenere forza muscolare e flessibilità, evitare sport ad alto impatto e lavori manuali pesanti, praticare nuoto per preservare la mobilità delle articolazioni per quanto possibile, se necessaria protesizzazione per ginocchia, anca, e spalle, interventi chirurgici per calcoli renali o prostatici, sostituzioni valvolari, trattamento sostitutivo con ormone tiroideo.
Una restrizione dietetica molto drastica degli aminoacidi fenilalanina e tirosina potrebbe ridurre la produzione di HGA come avviene per i pazienti fenilchetonurici, che assumono meno di 5-6 g di proteine naturali al giorno per mantenere bassi i livelli di fenilalanina. Tale trattamento dietetico dovrebbe essere seguito molto strettamente, per il rischio in primo luogo di carenze aminoacidiche che comprometterebbero la crescita ottimale dell’individuo (7).
È stato proposto il trattamento cronico con vitamina C come antiossidante che, tuttavia, non riduce l’escrezione di HGA e non ha efficacia chiaramente dimostrata (6,7).
Il trattamento con nitisinone è stato recentemente approvato dall’Agenzia europea per i medicinali in seguito ad uno studio internazionale che ne ha dimostrato l’efficacia nel rallentare la progressione della malattia (11). Si discute sull’età in cui iniziarlo, naturalmente in relazione alla gravità dei segni e sintomi del singolo individuo e ai possibili effetti collaterali, se iniziare presto a trattare con una dose bassa, e se anche i pazienti con patologia avanzata possano trarne beneficio (12). Durante il trattamento deve inoltre essere ridotto l’apporto proteico per evitare che si alzi la tirosina plasmatica fino a raggiungere livelli simili a quelli della tirosinemia tipo II e causare lesioni corneali e cutanee (11). Nella sezione “Il farmaco” di questo numero, l’iter della sperimentazione e approvazione di nitisinone per l’AKU è descritto in dettaglio.
Bibliografia
- Mistry JB, Bukhari M, Taylor AM. Alkaptonuria. Rare Diseases. 2013, 1, e27475.
- Braconi D, Millucci L, Spiga O, Santucci, A. Cell and tissue models of alkaptonuria. Drug Discovery Today: Disease Models. 2020; 31, 3–10.
- Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. The New England journal of medicine. 2002;347, 2111–2121.
- Zatkova A, Ranganath L, Kadasi L. Alkaptonuria: Current Perspectives. The application of clinical genetics. 2020; 13, 37.
- Millucci L, Braconi D, Bernardini G, et al. Amyloidosis in alkaptonuria. Journal of inherited metabolic disease. 2015; 38, 797–805.
- Braconi D, Millucci L, Bernardini G, Santucci A. Oxidative stress and mechanisms of ochronosis in alkaptonuria. Free radical biology & medicine. 2015; 88, 70–80.
- de Haas V, Carbasius Weber EC, de Klerk JB, et al. The success of dietary protein restriction in alkaptonuria patients is age-dependent. Journal of inherited metabolic disease. 1998; 21, 791–798.
- Introne WJ, Perry M, Chen M. Alkaptonuria. 2003 May 9 [updated 2021 Jun 10]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® 1993.
- Braconi D, Giustarini D, Marzocchi B, et al. Inflammatory and oxidative stress biomarkers in alkaptonuria: data from the DevelopAKUre project. Osteoarthritis and Cartilage. 2018; 26, 1078–1086.
- Nemethova M, Radvanszky J, Kadasi L,et al. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: Focus on “black bone disease” in Italy. European journal of human genetics: EJHG. 2016; 24, 66–72.
- Ranganath LR, Psarelli EE, Arnoux JB, et al. Efficacy and safety of once-daily nitisinone for patients with alkaptonuria (SONIA 2): an international, multicentre, open-label, randomised controlled trial. The lancet. Diabetes & endocrinology. 2020;8, 762–772.
- Häberle J. Suitability of nitisinone for alkaptonuria. The lancet. Diabetes & endocrinology. 2020;8(9), 732-733.