




la Newsletter

Nicola Tovaglieri, Elena Altieri
UOC di Pediatria, ASST Grande Ospedale Metropolitano Niguarda Ca' Granda, Milano
Anemia di Diamond-Blackfan | La malattia si manifesta con grave...
La malattia si manifesta con grave anemia dalla nascita o dai...
 L’anemia di Diamond-Blackfan (DBA) è una rara aplasia congenita della serie eritroide che provoca una grave anemia normocromica-macrocitica. Descritta per la prima volta nel 1936 da Hugh W. Josephs, fu meglio caratterizzata nel 1938 da Louis K. Diamond e Kennet Blackfan, cui si deve l’eponimo.
L’anemia di Diamond-Blackfan (DBA) è una rara aplasia congenita della serie eritroide che provoca una grave anemia normocromica-macrocitica. Descritta per la prima volta nel 1936 da Hugh W. Josephs, fu meglio caratterizzata nel 1938 da Louis K. Diamond e Kennet Blackfan, cui si deve l’eponimo.
Epidemiologia
L’incidenza è 5-7 casi/1.000.000 di nati vivi. L’età media alla diagnosi è di 2-3 mesi, il 90% dei casi è diagnosticato nel primo anno di vita.
Manifestazioni cliniche
Le manifestazioni cliniche possono essere molto eterogenee. Nel 20% dei casi si verifica una nascita prematura con un ritardo di crescita intrauterino.
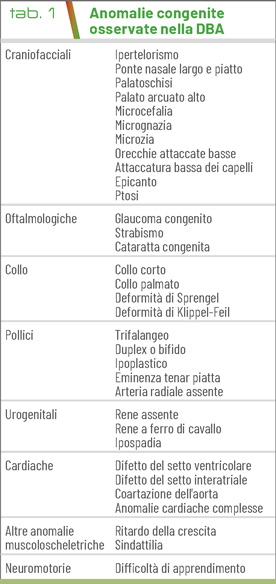
Nella prima infanzia si manifesta con i segni classici di anemia: pallore mucocutaneo, scarsa crescita, difficoltà nell’alimentazione. Più di un terzo dei pazienti presenta malformazioni tra cui: dismorfismi cranio-facciali (50% dei pazienti), anomalie degli arti superiori e in particolare del pollice (38%), malformazioni cardiache (30%) e urogenitali (39%), anomalie oculari, bassa statura e ritardo mentale (Tab. 1).
In circa il 50% dei pazienti sono presenti una o più endocrinopatie (insufficienza surrenalica, ipogonadismo, ipotiroidismo, disfunzione dell'ormone della crescita, diabete mellito e diabete insipido). Gli individui affetti presentano un aumentato rischio di sviluppo di patologie ematologiche (sindromi mielodisplastiche, leucemia acuta mieloide) e tumori solidi (carcinoma del colon, osteosarcoma).
Genetica e fisiopatologia
 La DBA è una ribosomopatia causata da mutazioni loss of function o delezioni che coinvolgono geni che codificano per proteine ribosomiali. Fino ad oggi 23 geni ribosomiali sono stati associati alla DBA, il più frequentemente coinvolto è RPS19, mutato nel 25% dei casi; altre mutazioni coinvolgono altri geni che codificano per la subunità grande (RPS26, RPS10 etc.) e la subunità piccola (RPL5, RPL11, RPL35A etc.) dei ribosomi. L’ereditarietà è autosomica dominante, le mutazioni in omozigosi di questi geni sono considerate letali. Molto più raramente sono state identificate mutazioni in altri geni come EPO e GATA1, con ereditarietà recessiva o X-linked e responsabili di forme non classiche di DBA (Fig. 1). La penetranza della DBA è incompleta e il fenotipo eterogeneo, da forme silenti a quadri moderato-severi.
La DBA è una ribosomopatia causata da mutazioni loss of function o delezioni che coinvolgono geni che codificano per proteine ribosomiali. Fino ad oggi 23 geni ribosomiali sono stati associati alla DBA, il più frequentemente coinvolto è RPS19, mutato nel 25% dei casi; altre mutazioni coinvolgono altri geni che codificano per la subunità grande (RPS26, RPS10 etc.) e la subunità piccola (RPL5, RPL11, RPL35A etc.) dei ribosomi. L’ereditarietà è autosomica dominante, le mutazioni in omozigosi di questi geni sono considerate letali. Molto più raramente sono state identificate mutazioni in altri geni come EPO e GATA1, con ereditarietà recessiva o X-linked e responsabili di forme non classiche di DBA (Fig. 1). La penetranza della DBA è incompleta e il fenotipo eterogeneo, da forme silenti a quadri moderato-severi.
Pur mancando una precisa correlazione genotipo-fenotipo, la palatoschisi e le anomalie dei pollici sono più frequentemente associate a mutazioni di RPL5 e RPL11; i pazienti con mutazioni di RPS24 e RPL11 hanno più probabilità di remissione, quelli con mutazioni di RPL5 e RPL11 di avere malformazioni congenite associate. I pazienti con DBA dovuta ad ampie delezioni in RPL35A hanno un fenotipo di malattia complesso e multisistemico con un’alta frequenza di anemia resistente agli steroidi, neutropenia, anomalie craniofacciali, problemi gastrointestinali cronici e disabilità intellettiva.
Il midollo dei pazienti affetti mostra un’aumentata apoptosi e un’alterata maturazione dei progenitori eritroidi, mentre le altre linee emopoietiche maturano normalmente.
 Diagnosi
Diagnosi
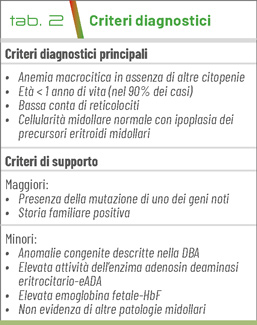
La presentazione classica dal punto di vista laboratoristico include severa anemia normo-macrocitica (MCV> 100 fl) e reticolocitopenia (<20.000/mmc) che si manifestano entro il primo anno di vita, senza altre significative citopenie (possibile neutropenia, moderata trombocitopenia, occasionalmente trombocitosi), e una assenza o presenza estremamente ridotta (<5%) di precursori eritroidi nel midollo osseo. L’emoglobina fetale è elevata dopo 6 mesi. L’aumento di concentrazione dell’eritropoietina è presente ma non specifico. L’adenosina deaminasi eritrocitaria (eADA) è elevata nel 75-90% dei casi di pazienti non trasfusi, con un valore predittivo negativo del 91%.
Elementi anamnestici, clinici e di laboratorio contribuiscono alla diagnosi: per la forma classica è necessaria la presenza dei 4 criteri diagnostici (Tab. 2).
Per la diagnosi di DBA atipica sono necessari 3 criteri diagnostici e 1 criterio di supporto maggiore, oppure 3 criteri diagnostici e 2 criteri di supporto minori, oppure 2 criteri diagnostici e 3 criteri di supporto minori, oppure 2 criteri di supporto maggiori.
Posta diagnosi di DBA, devono essere eseguiti alcuni esami di completamento diagnostico per identificare eventuali malformazioni associate: ecocardiogramma, ecografia addominale, Rx arti superiori.
Diagnosi differenziale
La diagnosi differenziale include una vasta gamma di patologie. Fra le forme acquisite, le infezioni virali (in particolare da Parvovirus B19 e HIV) e l’esposizione a farmaci e tossine, oltre ad alcune patologie immunomediate. Fra le forme congenite, devono essere considerate in particolare la sindrome di Shwachman-Diamond, quella di Pearson e l’anemia di Fanconi.
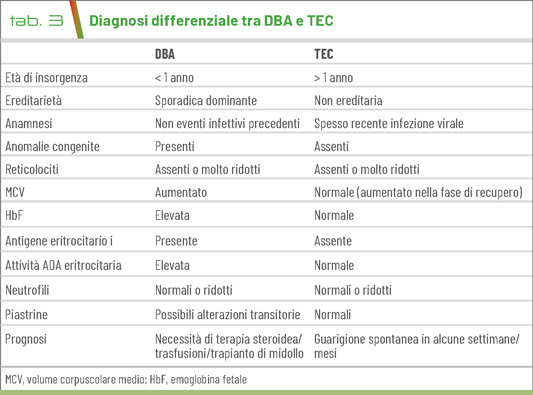 Nei bambini deve essere valutata con attenzione la possibilità dell’eritroblastopenia transitoria dell'infanzia (TEC) (Tab. 3).
Nei bambini deve essere valutata con attenzione la possibilità dell’eritroblastopenia transitoria dell'infanzia (TEC) (Tab. 3).
Trattamento
Le tre principali opzioni terapeutiche per l'anemia nella DBA sono le trasfusioni di globuli rossi concentrati, i glucocorticoidi e il trapianto allogenico di cellule ematopoietiche.
Nel 90% dei pazienti la necessità di trasfusioni si verifica prima del compimento dell’anno di età. La maggior parte dei pazienti necessita di 10-15 ml/kg di emazie ogni 3-5 settimane per mantenere valori di emoglobina compresi tra 8 e 9 mg/dL.
L’utilizzo di glucocorticoidi non è raccomandato prima dell’anno di età, e può essere ulteriormente procrastinato nei pazienti con grave ritardo di crescita.
Nei bambini di età maggiore all’anno i glucocorticoidi (prednisone) vengono avviati alla dose standard di 2 mg/kg per un periodo massimo di 4 settimane. Nei pazienti che rispondono al trattamento, l’aumento dei reticolociti è evidente in circa 10 giorni e il livello di emoglobina raggiunge valori normali in circa 1 mese. Nessuna evidenza supporta il trattamento prolungato a questi dosaggi, pertanto dopo l’aumento dei reticolociti il dosaggio viene gradualmente ridotto fino alla dose minima necessaria per mantenere adeguati livelli di emoglobina. Circa il 60% dei pazienti risponde agli steroidi con una risposta completa (remissione della malattia anche senza terapia) o duratura ma dipendente dalla terapia stessa (steroido-dipendenza), a dosaggio variabile. Nei Paesi con adeguata disponibilità di sangue la massima dose tollerata di steroidi è 0,3 mg/kg/die (0,5 mg/Kg/die invece nei Paesi con scarsa disponibilità di sangue); i pazienti con necessità di dosi maggiori vengono inseriti in programmi di trasfusione periodica.
Durante il trattamento sono raccomandati la supplementazione con vitamina D ed il controllo periodico della densitometria ossea. La sospensione degli steroidi si considera in caso di ritardo di crescita nei pazienti pre-adolescenti, durante la gravidanza e quando si verificano severi effetti collaterali iatrogeni. Il 35% circa dei pazienti è steroido-resistente.
Per i pazienti trasfusione dipendenti il rischio maggiore è il sovraccarico marziale.
In Europa la chelazione viene iniziata prima dei 2 anni con deferoxamina, in genere dopo 10-20 trasfusioni anche in relazione all’accumulo marziale (cut off ferritina > 1000 μg/L e/o saturazione della transferrina > 75%, ferro epatico > 6-7 mg/g). Nei bambini sopra i 2 anni di età può essere invece usato deferasirox, se necessario in associazione con deferoxamina. L’obiettivo è mantenere i livelli di ferritina <500 μg/L e normale il ferro epatico alla risonanza magnetica.
La terapia con glucocorticoidi ed il sovraccarico marziale possono essere una concausa di endocrinopatia in pazienti affetti da DBA.
Il trapianto allogenico di cellule ematopoietiche è l’unica opzione curativa e deve essere considerato precocemente (3-5 anni) nei pazienti trasfusione dipendenti. Le indicazioni sono l’assenza di risposta alla terapia steroidea, la tossicità da steroidi, il fallimento della chelazione e la severa neutropenia associata. Il trapianto deve essere invece considerato con cautela negli adolescenti e negli adulti senza evoluzione clonale, perché, seppur riduca il rischio di sindromi mielodisplastiche e leucemie, può aumentare il rischio di tumori solidi.
La terapia genica è una potenziale opzione terapeutica: vettori lentivirali si sono mostrati promettenti nel trattamento delle DBA da mutazione RPS19 promuovendo una normale ematopoiesi.
Per il ritardo di crescita la terapia con l'ormone della crescita (GH) si è dimostrata essere efficace, seppur vada considerata la predisposizione allo sviluppo di tumori.
Conclusioni
L’anemia di Diamond-Blackfan è una rara causa di anemia che deve essere sospettata in pazienti con anemia normo-macrocitica iporigenerativa, soprattutto se associata a tratti fenotipici peculiari o a familiarità per anemia.
L’iter diagnostico comprende la valutazione di esame emocromocitometrico con reticolociti e dosaggio dell’emoglobina fetale, successivamente dosaggio di eritropoietina, adenosina deaminasi, esame morfologico di sangue midollare, studio delle subunità ribosomali e ricerca di mutazioni genetiche note. Parallelamente devono essere escluse forme di eritroblastopenia transitoria (per lo più legate ad infezioni virali) ed insufficienza midollare. Nei pazienti trasfusione dipendenti o con elevato fabbisogno di steroidi il trapianto di midollo osseo deve essere considerato precocemente. I pazienti affetti devono essere monitorati nel tempo anche per il rischio di sviluppo di patologie maligne.
In considerazione della estrema variabilità clinica e laboratoristica, è all’esame di un panel di esperti una modifica della denominazione “anemia di Diamond-Blackfan” in “sindrome di Diamond-Blackfan” con una parziale revisione dei criteri diagnostici, che a breve verrà pubblicata.
Bibliografia
- Da Costa L, Leblanc T, Mohandas N. Diamond-Blackfan anemia. Blood. 2020;136(11):1262-1273.
- Liu Y, Karlsson S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia. 2024;38(1):1-9. Erratum in: Leukemia. 2023 Dec 13; PMID: 37973818; PMCID: PMC10776401.
- Bhoopalan SV, Suryaprakash S, Sharma A, Wlodarski MW. Hematopoietic cell transplantation and gene therapy for Diamond-Blackfan anemia: state of the art and science. Front Oncol. 2023;13:1236038.
- Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859-76.
- Lahoti A, Harris YT, Speiser PW, et al. Endocrine Dysfunction in Diamond-Blackfan Anemia (DBA): A Report from the DBA Registry (DBAR). Pediatr Blood Cancer. 201663(2):306-12.