




la Newsletter

Chiara Parazzoli1, Vittoria Favero1, Iacopo Chiodini1,2
1Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano; 2Struttura Complessa di Endocrinologia, ASST Ospedale Niguarda, Milano
Feocromocitoma: manifestazioni sindromiche | Da oltre un secolo...
Da oltre un secolo sono state documentate le caratteristiche...
Feocromocitoma e paraganglioma
Il feocromocitoma e il paraganglioma (PPGL) sono tumori neuroendocrini rari che si sviluppano rispettivamente a partire dal tessuto cromaffine della midollare del surrene e dai paragangli extra-surrenalici del sistema nervoso simpatico e parasimpatico. I paragangliomi che originano dai gangli parasimpatici sono generalmente non funzionanti e sono localizzati a livello del collo e/o della testa; diversamente i paragangliomi che originano dai gangli del sistema nervoso simpatico si localizzano in sede addominale e/o toracica e spesso producono catecolamine.
L'incidenza di questi tumori è di circa 0,6 casi per 100.000 persone all'anno. In passato, si pensava che circa il 10% di questi tumori fosse legato a malattie ereditarie come la neoplasia endocrina multipla tipo 2 (MEN2), la sindrome di von Hippel-Lindau (VHL) e la neurofibromatosi di tipo 1 (NF1). Tuttavia, con l'identificazione progressiva di nuove mutazioni genetiche (SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, MAX, FH, MDH2), si stima che almeno il 30% di essi sia causato da una mutazione genetica ereditaria. Pertanto, le linee guida attuali raccomandano che tutti i pazienti con diagnosi di PPGL siano sottoposti a consulenza genetica per valutare se sia indicato sottoporsi alle indagini per la ricerca di mutazioni genetiche.
Gestione clinica
I pazienti con PPGL possono manifestare una vasta gamma di sintomi, che tradizionalmente sono definiti dalla triade: cefalea, palpitazioni e sudorazione intensa. Tuttavia, a causa dell'uso sempre più diffuso di tecniche diagnostiche per immagini, sta diventando sempre più comune identificare il feocromocitoma come incidentaloma surrenalico. Inoltre, sono sempre più frequenti i casi di PPGL asintomatici che vengono diagnosticati in base alla storia familiare e alla presenza di mutazioni ereditarie.
Per la diagnosi di feocromocitoma o paraganglioma sono necessari la dimostrazione di un rilascio eccessivo di catecolamine e la conferma all’imaging del tumore. L'aumento dei livelli di metanefrine frazionate (metanefrina e normetanefrina) nel plasma è un marker sensibile e specifico, con una sensibilità del 97% e una specificità del 93% (Fig. 1).

Attualmente, la misurazione delle metanefrine frazionate nel plasma è poco diffusa, mentre molti laboratori consentono la misurazione delle metanefrine urinarie, che ha analoga sensibilità ma inferiore specificità, con una quota non trascurabile di falsi positivi. In generale, comunque, livelli lievemente elevati di metanefrine e catecolamine possono essere riscontrati anche in individui sani.
L'imaging funzionale, come la scintigrafia con 123I-MIBG o la PET-CT con DOTATATE o l-DOPA, è altamente efficace nella localizzazione di feocromocitomi e paragangliomi. Le principali indicazioni per l'imaging funzionale comprendono la ricerca di malattia metastatica o l'identificazione di tumori multipli.
I paragangliomi del collo e della testa di solito si presentano come masse asintomatiche a crescita lenta, principalmente localizzate nel corpo carotideo e nei paragangliomi vagali. Nei pazienti con forme avanzate di paragangliomi del collo e della testa, sono comuni lievi disfunzioni dei nervi cranici e può essere presente perdita dell’udito. L'ipersecrezione di catecolamine è rara in questi pazienti.
L'intervento chirurgico rappresenta il punto cardine nella terapia del feocromocitoma. La scelta del trattamento per pazienti affetti da paraganglioma testa-collo richiede un approccio personalizzato e interdisciplinare. Tra le opzioni terapeutiche disponibili vi sono interventi chirurgici, radiochirurgia, radioterapia e una strategia di monitoraggio.
Sindromi associate al feocromocitoma
Da oltre un secolo sono state documentate le caratteristiche cliniche delle sindromi correlate al feocromocitoma, tra cui la NF1, la VHL e la MEN2. Con l'identificazione dei geni di suscettibilità e delle varie mutazioni germinali, queste sindromi sono state meglio definite e caratterizzate. Il 50% dei pazienti affetti da queste sindromi sviluppa feocromocitomi unilateralmente o bilateralmente.
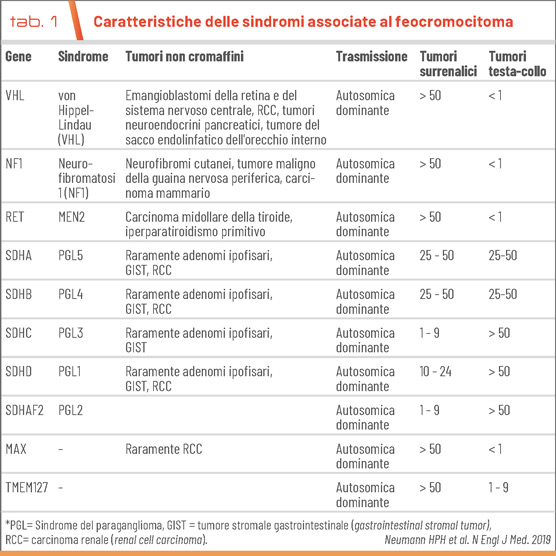 Oltre alla storia familiare, i segni tipici di feocromocitoma e paraganglioma ereditari includono un'età precoce di insorgenza, tumori primari extrasurrenalici e multipli, e tumori non paragangliari associati. L'età alla diagnosi dei feocromocitomi e dei paragangliomi sindromici è di circa 15 anni più giovane rispetto ai casi sporadici. La presenza di un paraganglioma in una sede insolita, come l'organo di Zuckerkandl, il torace o la vescica, dovrebbe far sospettare una sindrome associata al tumore. Le sedi anatomiche dei feocromocitomi e dei paragangliomi differiscono notevolmente tra le diverse sindromi (Tab. 1).
Oltre alla storia familiare, i segni tipici di feocromocitoma e paraganglioma ereditari includono un'età precoce di insorgenza, tumori primari extrasurrenalici e multipli, e tumori non paragangliari associati. L'età alla diagnosi dei feocromocitomi e dei paragangliomi sindromici è di circa 15 anni più giovane rispetto ai casi sporadici. La presenza di un paraganglioma in una sede insolita, come l'organo di Zuckerkandl, il torace o la vescica, dovrebbe far sospettare una sindrome associata al tumore. Le sedi anatomiche dei feocromocitomi e dei paragangliomi differiscono notevolmente tra le diverse sindromi (Tab. 1).
Sindrome MEN2 (Multiple Endocrine Neoplasia)
La MEN2 è causata da mutazioni attivanti il protoncogene RET che codifica per un recettore transmembrana appartenente alla famiglia delle tirosin-chinasi, coinvolto nella regolazione dell’apoptosi e della proliferazione cellulare.
La MEN2 comprende 3 sottotipi clinici: MEN2A, MEN2B e carcinoma midollare familiare della tiroide.
Una volta identificata una mutazione patogenica del gene RET in un paziente con feocromocitoma, i clinici devono essere consapevoli che la quasi totalità dei portatori della mutazione svilupperà il carcinoma midollare della tiroide, mentre il 20% dei pazienti con MEN2A, ma non con MEN2B, svilupperà iperparatiroidismo.
Tuttavia, è raro che un paziente con MEN2B presenti il feocromocitoma come prima manifestazione, poiché le altre caratteristiche tipiche si manifestano più precocemente e spesso già in età infantile. Tra queste ricordiamo in particolare:
- il carcinoma midollare della tiroide
- i ganglioneuromi, che interessano tipicamente lingua, labbra e palpebre
- le deformità scheletriche (ad esempio, cifoscoliosi e habitus marfanoide)
- la lassità articolare
- la ganglioneuromatosi intestinale.
Sindrome di von Hippel-Lindau (VHL)
Si tratta di una malattia genetica causata da una mutazione del gene oncosoppressore VHL, con un'incidenza di 1 su 36.000 nati vivi. Essa è caratterizzata dalla formazione di lesioni cistiche e tumori benigni e maligni in diversi organi del corpo. Tra questi, vi sono gli emangioblastomi retinici e del SNC (cerebrali e spinali) e i tumori neuroendocrini del pancreas. Nei pazienti con VHL, in particolare quelli con tipo 2 della malattia, i feocromocitomi e i paragangliomi sono comuni e possono essere la manifestazione iniziale della malattia, anche in età pediatrica.
Neurofibromatosi tipo 1 (NF1) o malattia di von Recklinghausen
La NF1 ha un’incidenza di 1/3.000 persone ed è caratterizzata da neurofibromi cutanei e/o delle mucose, macchie caffelatte, lentiggini ascellari, amartomi iridei (noduli di Lisch), anomalie ossee, gliomi del SNC, macrocefalia e deficit cognitivi. I feocromocitomi e paragangliomi sono presenti solo nell'1-3% delle persone colpite.
Sindromi del Paraganglioma Familiare
Causate da mutazioni nei geni SDHx, queste sindromi sono ereditate in modo autosomico dominante, anche se la penetranza incompleta può influenzare la manifestazione clinica. Più in dettaglio, i geni SDHD e SDHAF2 mostrano un imprinting materno, quindi i sintomi si manifestano solo se l'allele mutato viene trasmesso dal padre.
I geni SDHx codificano per le proteine del complesso II della succinato-deidrogenasi, enzima coinvolto nella catena di trasporto degli elettroni nel mitocondrio. I pazienti con mutazioni SDHx generalmente sviluppano feocromocitomi e paragangliomi. In una minoranza di portatori di mutazioni di SDHx sono stati osservati tumori stromali gastrointestinali (GIST), carcinomi a cellule renali o adenomi ipofisari.
La sindrome autosomica dominante di Carney-Stratakis è caratterizzata dall'associazione di feocromocitoma, paraganglioma o di entrambi con GIST. La cosiddetta sindrome delle 3PAs (feocromocitoma, paraganglioma e adenoma ipofisario) è associata alle mutazioni di SDHx.
I pazienti con mutazioni di TMEM127 o MAX hanno generalmente solo feocromocitomi e paragangliomi, anche se la variante mutata del gene TMEM127 è stata invece riscontrata in una piccola percentuale di soggetti con carcinoma renale, così come la mutazione del gene MAX è stata riportata in pazienti con oncocitoma renale.
Bibliografia
- Lenders JWM, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-42.
- Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and Paraganglioma. N Engl J Med. 2019;381(6):552-565.
- Fassnacht M, Tsagarakis S, Terzolo M, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2023;189(1):G1-G42.
- Muth A, Crona J, Gimm O, et al. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med. 2019; 285:187–204.
- Calissendorff J, Juhlin CC, Bancos I, Falhammar H. Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance. Cancers. 2022;14(4): 917.