




la Newsletter

Alessandro Plebani
Clinica Pediatrica, Dipartimento di Scienze Cliniche e Sperimentali, Università degli Studi di Brescia-ASST Spedali Civili di Brescia
IPINet, il registro italiano delle immunodeficienze primitive | La...
La rete è nata per definire la distribuzione e le caratteristiche...
Un gruppo eterogeneo di patologie
Le immunodeficienze primitive (IDP) sono malattie rare congenite caratterizzate da un difetto nei meccanismi di difesa del sistema immunitario. Pertanto l’aumentata suscettibilità alle infezioni, che possono essere anche mortali, rappresenta la caratteristica principale di queste malattie. La gravità delle infezioni varia a seconda del tipo di immunodeficienza. Le IDP sono classificate in immunodeficienze umorali (difetto numerico o funzionale dei linfociti B: agammaglobulinemia, ipogammaglobulinemia comune variabile, immunodeficienza con Iper IgM, deficit selettivo di IgA), immunodeficienze cellulari (difetto numerico o funzionale dei linfociti T: immunodeficienze combinate) e immunodeficienze da difetti numerici o funzionali dei granulociti (neutropenie e malattia granulomatosa cronica). Vi sono poi forme di immunodeficienza in cui il difetto immunologico fa parte di un quadro più complesso che configura nell’insieme una sindrome (immunodeficienze sindromiche: sindrome di DiGeorge, atassia-teleangiectasia, sindrome di Wiskott Aldrich, ecc.). Per una classificazione più dettagliata si rimanda al recente lavoro di Tangye e colleghi (1). Le immunodeficienze combinate rappresentano le forme di IDP più gravi per via della aumentata suscettibilità a severe infezioni da patogeni opportunisti e il trapianto di midollo osseo rappresenta il trattamento di elezione. Per le forme umorali il trattamento di prima scelta è la terapia sostitutiva con immunoglobuline, mentre per le forme sindromiche varia a seconda della complessità del quadro clinico. In ogni caso una diagnosi precoce ed un trattamento adeguato e tempestivo rappresentano i cardini terapeutici più importanti per queste malattie.
IPINet, il registro delle IDP
 Le IDP sono malattie rare e come tali non sempre adeguatamente conosciute anche dalla classe medica e dai servizi socio-sanitari. In passato, spesso, i pazienti venivano seguiti da più figure specialistiche (gastroenterologi, allergologi, reumatologi, pneumologi) per via della variabilità dei sintomi di presentazione o ricorrenti, ma non erano presi in carico da esperti di immunodeficienze primitive, figure che peraltro scarseggiavano. Infatti, soltanto pochi centri sul territorio nazionale erano altamente specializzati nella diagnosi e nel trattamento di queste malattie. Inoltre, il passaggio di competenze del sistema sanitario alle 20 Regioni rendeva ancora più difficile garantire ai pazienti le dovute prestazioni assistenziali ed il follow-up da parte di centri specializzati extra-regionali. Tutto questo comportava inevitabilmente un ritardo diagnostico e terapeutico, e non di rado, un trattamento non adeguato. Tra l’altro, una volta formulata la diagnosi, i pazienti e i loro familiari provavano un inevitabile senso di smarrimento ed isolamento nel gestire malattie così complesse e poco note anche alle istituzioni sanitarie. Forte era la loro richiesta di una condivisione delle procedure diagnostiche, terapeutiche e dei servizi tra i vari ospedali/servizi socio-sanitari del territorio nazionale che seguivano questi pazienti.
Le IDP sono malattie rare e come tali non sempre adeguatamente conosciute anche dalla classe medica e dai servizi socio-sanitari. In passato, spesso, i pazienti venivano seguiti da più figure specialistiche (gastroenterologi, allergologi, reumatologi, pneumologi) per via della variabilità dei sintomi di presentazione o ricorrenti, ma non erano presi in carico da esperti di immunodeficienze primitive, figure che peraltro scarseggiavano. Infatti, soltanto pochi centri sul territorio nazionale erano altamente specializzati nella diagnosi e nel trattamento di queste malattie. Inoltre, il passaggio di competenze del sistema sanitario alle 20 Regioni rendeva ancora più difficile garantire ai pazienti le dovute prestazioni assistenziali ed il follow-up da parte di centri specializzati extra-regionali. Tutto questo comportava inevitabilmente un ritardo diagnostico e terapeutico, e non di rado, un trattamento non adeguato. Tra l’altro, una volta formulata la diagnosi, i pazienti e i loro familiari provavano un inevitabile senso di smarrimento ed isolamento nel gestire malattie così complesse e poco note anche alle istituzioni sanitarie. Forte era la loro richiesta di una condivisione delle procedure diagnostiche, terapeutiche e dei servizi tra i vari ospedali/servizi socio-sanitari del territorio nazionale che seguivano questi pazienti.
.jpg) Ecco quindi che nel Novembre 1999 nasce la rete IPINet (Italian Primary Immunodeficiency network) fortemente voluta, oltre che dai medici anche dai pazienti e dai loro familiari riuniti sotto l’egida della Associazione Immunodeficienze Primitive (AIP). Gli obiettivi che IPINet si proponeva erano i seguenti:
Ecco quindi che nel Novembre 1999 nasce la rete IPINet (Italian Primary Immunodeficiency network) fortemente voluta, oltre che dai medici anche dai pazienti e dai loro familiari riuniti sotto l’egida della Associazione Immunodeficienze Primitive (AIP). Gli obiettivi che IPINet si proponeva erano i seguenti:
- informare i medici sulle IDP e su come riconoscerle
- mappare le IDP sul territorio nazionale
- fornire a tutti i bambini italiani i migliori strumenti diagnostici e terapeutici e la possibilità di venire curati in un ospedale vicino a casa seguendo protocolli di cura aggiornati e condivisi
- studiare l’evoluzione della malattia per ridurre la frequenza e la gravità delle complicanze e per prolungare la sopravvivenza con una miglior qualità di vita.
Gli strumenti utilizzati
 Da qui la necessità di far conoscere queste malattie in vari ambiti del territorio nazionale: nelle diverse specializzazioni mediche, nella medicina pubblica e nel personale paramedico e di assistenza oltre che nel sociale. Obiettivo perseguito mediante incontri annuali patrocinati dall’AIP in diverse città d’Italia. Partecipavano a questi incontri medici di tutto il territorio nazionale esperti in immunodeficienze primitive, medici ospedalieri e della città/regione dove si svolgeva l’incontro e rappresentanti delle istituzioni sanitarie locali.
Da qui la necessità di far conoscere queste malattie in vari ambiti del territorio nazionale: nelle diverse specializzazioni mediche, nella medicina pubblica e nel personale paramedico e di assistenza oltre che nel sociale. Obiettivo perseguito mediante incontri annuali patrocinati dall’AIP in diverse città d’Italia. Partecipavano a questi incontri medici di tutto il territorio nazionale esperti in immunodeficienze primitive, medici ospedalieri e della città/regione dove si svolgeva l’incontro e rappresentanti delle istituzioni sanitarie locali.
Non di rado dagli incontri emergevano differenze regionali nella gestione clinico-assistenziale dei pazienti evidenziando situazioni di “disuguaglianza”. Si poneva quindi la necessità di rendere omogenea a livello nazionale l’assistenza ai pazienti secondo protocolli di diagnosi e terapia formulati sulla base di consensi internazionali. Criticità risolta con la messa on line di protocolli condivisi ed applicati dai 60 ospedali del territorio nazionale che nel frattempo avevano aderito alla rete, garantendo così l’assistenza più aggiornata presso il centro più vicino alla residenza del paziente (https://www.aieop.org/web/operatori-sanitari/gruppi-di-lavoro/immunodeficienze/).
Venivano evitati in questo modo spostamenti ad altri centri, anche molto distanti, e notevoli disagi economici, familiari e organizzativi. Nello stesso tempo, mediante la rete ci si è proposti, utilizzando i sistemi informatici del CINECA attraverso l’AIEOP (Associazione Italiana di Emato-Oncologia Pediatrica), di creare una banca dati sulla distribuzione dei pazienti con le varie forme di IDP sul territorio nazionale, di costruire, attraverso un aggiornamento biennale dei dati clinici, la storia naturale di queste malattie, utile per elaborare strategie preventive e terapeutiche atte a ridurre la frequenza e la gravità delle complicanze e a prolungare la sopravvivenza con una miglior qualità di vita.
I risultati
Nel giro di pochi anni i benefici di questa rete non hanno tardato a dare i loro frutti (2):
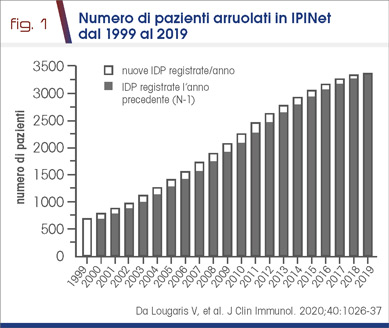
- il numero dei pazienti con IDP registrati nella rete IPINet è andato progressivamente aumentando come conseguenza di una maggiore conoscenza da parte della classe medica (Fig.1)
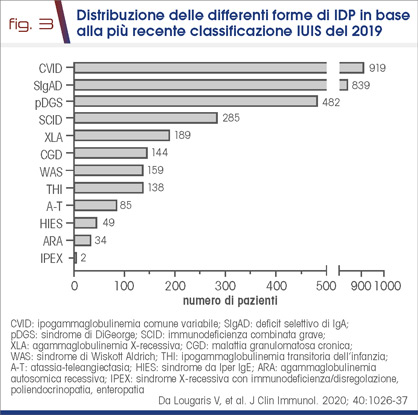
- si è potuto mappare sul territorio nazionale la distribuzione per regione di queste malattie (Fig. 2) oltre che la tipologia delle varie forme di IDP (Fig. 3)
- l’applicazione di protocolli diagnostici e terapeutici ha consentito di pervenire ad una diagnosi più precoce e quindi di instaurare un trattamento più tempestivo, riducendo il tasso di mortalità
- il follow-up a lungo termine di questi pazienti ha permesso di identificare le complicanze tardive e di sviluppare strategie terapeutiche preventive più efficaci. L’agammaglobulinemia ne rappresenta un esempio. Nonostante il trattamento sostitutivo con immunoglobuline endovena o sottocutaneo costituisca la terapia elettiva di questa immunodeficienza, questo trattamento non è in grado di controllare in modo ottimale le infezioni del tratto respiratorio, da qui lo sviluppo di una pneumopatia cronica che rappresenta tuttora la causa di maggiore mortalità di questi pazienti (3). Questa osservazione ha portato a considerare l’importanza di un adeguato trattamento fisioterapico, eventualmente associato ad antibioticoprofilassi.
Diagnosi precoce quindi, trattamento tempestivo e adeguato, secondo le più recenti raccomandazioni internazionali, e diffusione delle informazioni su come riconoscere queste malattie attraverso incontri sul territorio nazionale, rivolti ai pediatri e ai medici di medicina generale, hanno rappresentato gli elementi vincenti per ridurre la mortalità e migliorare la qualità di vita dei pazienti.
La rete IPINet ci ha insegnato, attraverso i periodici incontri di aggiornamento, a lavorare insieme, a condividere e confrontare esperienze assistenziali differenti e a modificarle, se del caso, nell’ottica di una migliore assistenza ai pazienti e ai loro familiari.
Bibliografia
- Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity:2019 update on the classification from the International Union of Immunological Societies Expert Commitee. J Clin Immunol. 2020; 40:24-64.
- Lougaris V, Pession A, Baronio E, et al. The Italian registry for primary immunodeficiencies (Italian Primary Immunodeficiency Network; IPINet):twenty years of esperience (1999-2019). J Clin Immunol. 2020; 40:1026-37.
- Lougaris V, Soresina A, Baronio M, et al. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol. 2020;146:429-37.