




la Newsletter

Jan Walter Schroeder, Linda Borgonovo
S.C. Allergologia e Immunologia, Dipartimento Medico Polispecialistico, ASST Grande Ospedale Metropolitano Niguarda, Milano
La granulomatosi eosinofila con poliangioite | Per questa...
Per questa patologia è particolarmente importante la...
.jpg) La granulomatosi eosinofila con poliangioite (EGPA), precedentemente nota come sindrome di Churg-Strauss, è una vasculite necrotizzante sistemica coinvolgente vasi sanguigni di piccolo e medio calibro ed è caratterizzata da eosinofilia periferica, asma, rinosinusite con o senza polipi, e interessamento di altri organi.
La granulomatosi eosinofila con poliangioite (EGPA), precedentemente nota come sindrome di Churg-Strauss, è una vasculite necrotizzante sistemica coinvolgente vasi sanguigni di piccolo e medio calibro ed è caratterizzata da eosinofilia periferica, asma, rinosinusite con o senza polipi, e interessamento di altri organi.
L'EGPA è classificata tra le cosiddette vasculiti sistemiche ANCA-associate (autoanticorpi anti citoplasma neutrofilo), anche se la positività agli ANCA è presente solo nel 30-40% dei pazienti. Gli ANCA, generalmente della variante p-ANCA (mieloperossidasi), possono aiutare a distinguere due differenti subset all’interno della EGPA. I pazienti con EGPA ANCA positivi presentano frequentemente manifestazioni come la mononeurite multipla, la glomerulonefrite a semilune e la porpora cutanea. Invece, i pazienti ANCA negativi presentano più frequentemente infiltrati polmonari, poliposi nasale, miocarditi e pleuriti. Altre manifestazioni cliniche come i sintomi sistemici (febbre, astenia, artromialgie e calo ponderale) hanno una distribuzione simile nei due sottotipi di EGPA.
 I criteri classificativi sono stati approvati nel 1990 dall’American College of Rheumatology (Tab. 1) e aggiornati nel 2022 (Tab. 2).
I criteri classificativi sono stati approvati nel 1990 dall’American College of Rheumatology (Tab. 1) e aggiornati nel 2022 (Tab. 2).
Aspetti epidemiologici ed eziologici
L’EGPA, malattia rara, presenta tassi di incidenza e prevalenza tra i più bassi rispetto alle altre vasculiti sistemiche. Per quanto riguarda l’Europa, i dati del French Vasculitis Study Group dimostrano una prevalenza che varia da 10,7 a 13 casi per milione di abitanti, con un’incidenza pari a 0,5-0,8 nuovi casi per anno su un milione di abitanti. L’insorgenza può verificarsi in qualsiasi periodo della vita, sebbene l’esordio sia più comune in età adulta, con una media attorno ai 50 anni. I casi diagnosticati in età pediatrica sono molto rari.
Attualmente l’eziologia dell’EGPA rimane ancora sconosciuta. Non è quindi chiara la causa che porta all’innesco dell’infiammazione eosinofilica e all’attivazione non controllata del sistema immunitario con una tempesta citochinica. Accanto a una predisposizione genetica, si pensa che fattori ambientali, infezioni virali o l’utilizzo di determinati farmaci, possano giocare un ruolo trigger nell’attivazione della patologia.
 Dal punto di vista fisiopatologico si osserva la comparsa di una vasculite innescata dagli ANCA e di un danno organico secondario all’infiltrazione tissutale degli eosinofili, con conseguente degranulazione e rilascio delle proteine [proteina cationica eosinofila (ECP), proteina maggiore basica (MBP), etc.] che provocano danni a livello dei tessuti e della circolazione, anche con eventi trombotici.
Dal punto di vista fisiopatologico si osserva la comparsa di una vasculite innescata dagli ANCA e di un danno organico secondario all’infiltrazione tissutale degli eosinofili, con conseguente degranulazione e rilascio delle proteine [proteina cationica eosinofila (ECP), proteina maggiore basica (MBP), etc.] che provocano danni a livello dei tessuti e della circolazione, anche con eventi trombotici.
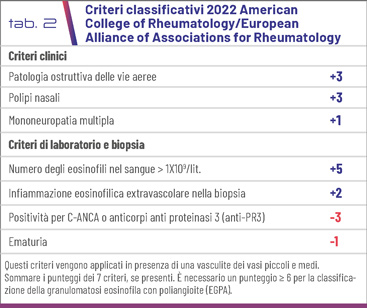
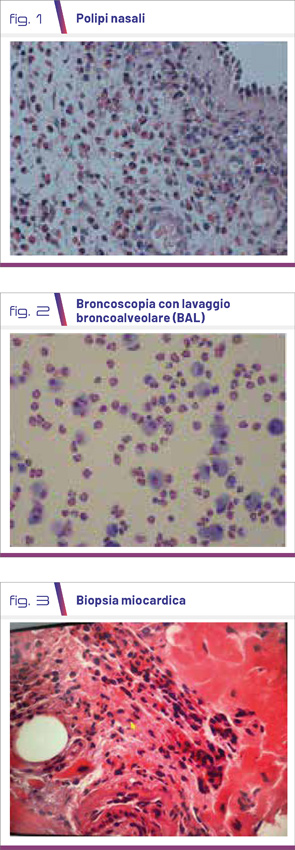
Si tratta di una malattia autoimmune che presenta tre fasi: la fase prodromica, caratterizzata da rinosinusite e asma, che evolve, dopo un periodo variabile da alcuni mesi fino a 8-10 anni, nella fase eosinofilica, dominata da infiltrati eosinofili a livello tissutale e granulomi eosinofili extravascolari (Fig. 1-3), infiltrati polipi, interessamento polmonare e cardiomiopatia eosinofila, per arrivare alla fase vasculitica con importanti sintomi sistemici e interessamento organico (p.es. glomerulonefrite, neuropatia del sistema nervoso periferico e porpora palpabile).
Quadri clinici
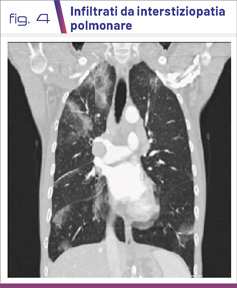
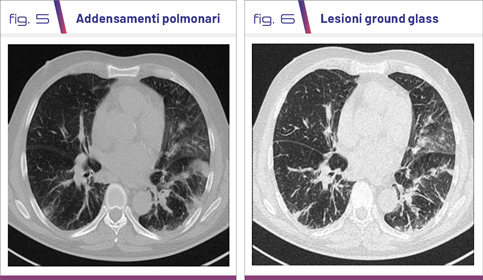
I pazienti si presentano all’esordio della malattia con sintomi sistemici importanti come febbre, calo ponderale, artralgie, mialgie e astenia profonda associati a sintomi respiratori come tosse stizzosa, dispnea ingravescente fino all’insufficienza respiratoria. L’asma è presente nella quasi totalità dei pazienti con prevalente esordio in età adulta (late onset). A livello toracico si riscontrano spesso addensamenti polmonari multipli come da interstiziopatia (lesioni ground glass) (Fig. 4-6).
 Sintomi da rinosinusite sono presenti nell’85% dei pazienti e solo nel 30% si tratta di una forma atopica. Una parte consistente presenta polipi nasali che recidivano precocemente dopo l’intervento chirurgico. Possono associarsi altre manifestazioni gravi come una mononeuropatia multipla con coinvolgimento degli arti superiori e inferiori che si manifesta con parestesie, dolori intensi, ipostenia fino all’impossibilità di camminare o muovere le mani (tetraparesi). Il coinvolgimento neurologico può interessare anche il sistema nervoso centrale con ischemia cerebrale da trombosi di vasi cerebrali oppure da embolia a partenza cardiaca. Altre manifestazioni riguardano il cuore che può essere coinvolto nel 30% dei pazienti con una miocardite eosinofila acuta da infiltrato eosinofilico, caratterizzata da un quadro clinico da insufficienza cardiaca fino allo shock cardiaco. In queste situazioni si manifestano anche trombi intracavitari che rappresentano un rischio per l’embolizzazione cerebrale. Inoltre si riconoscono spesso pericarditi con versamento e scollamento pericardico. Si associano frequentemente anche versamenti pleurici. Ci sono quadri da infarto miocardico da trombosi eosinofila coronarica oppure da spasmo coronarico. I reni sono coinvolti maggiormente nei pazienti che presentano anche gli ANCA con insufficienza renale da glomerulonefrite.
Sintomi da rinosinusite sono presenti nell’85% dei pazienti e solo nel 30% si tratta di una forma atopica. Una parte consistente presenta polipi nasali che recidivano precocemente dopo l’intervento chirurgico. Possono associarsi altre manifestazioni gravi come una mononeuropatia multipla con coinvolgimento degli arti superiori e inferiori che si manifesta con parestesie, dolori intensi, ipostenia fino all’impossibilità di camminare o muovere le mani (tetraparesi). Il coinvolgimento neurologico può interessare anche il sistema nervoso centrale con ischemia cerebrale da trombosi di vasi cerebrali oppure da embolia a partenza cardiaca. Altre manifestazioni riguardano il cuore che può essere coinvolto nel 30% dei pazienti con una miocardite eosinofila acuta da infiltrato eosinofilico, caratterizzata da un quadro clinico da insufficienza cardiaca fino allo shock cardiaco. In queste situazioni si manifestano anche trombi intracavitari che rappresentano un rischio per l’embolizzazione cerebrale. Inoltre si riconoscono spesso pericarditi con versamento e scollamento pericardico. Si associano frequentemente anche versamenti pleurici. Ci sono quadri da infarto miocardico da trombosi eosinofila coronarica oppure da spasmo coronarico. I reni sono coinvolti maggiormente nei pazienti che presentano anche gli ANCA con insufficienza renale da glomerulonefrite.
 Il tratto gastrointestinale è meno frequentemente coinvolto ma i pazienti presentano spesso dolori addominali, alterazioni dell’alvo e calo ponderale. Si possono riscontrare ulcere coliche e perforazione intestinale. A livello cutaneo può comparire una porpora palpabile ma anche orticaria o noduli cutanei che istologicamente evidenziano infiltrati eosinofili con segni di vasculite che, se presenti, favoriscono la diagnosi.
Il tratto gastrointestinale è meno frequentemente coinvolto ma i pazienti presentano spesso dolori addominali, alterazioni dell’alvo e calo ponderale. Si possono riscontrare ulcere coliche e perforazione intestinale. A livello cutaneo può comparire una porpora palpabile ma anche orticaria o noduli cutanei che istologicamente evidenziano infiltrati eosinofili con segni di vasculite che, se presenti, favoriscono la diagnosi.
Diagnosi
La diagnosi deve essere formulata rapidamente perché la malattia può avere un decorso molto veloce e violento verso la fase vasculitica. Generalmente si tratta di persone con storia di asma, rinosinusite cronica, ipereosinofilia ematica (≥1.500/μl) nei quali compaiono sintomi acuti sistemici o a carico di un organo. I valori degli eosinofili possono essere molto alti (fino a 20-30.000) come anche gli indici infiammatori (VES e PCR). Si riscontra spesso anche un aumento degli IgE totali, anche nei pazienti non allergici e aumenti dell’LDH e della beta-2-microglobulina. Nel 30% dei pazienti sono presenti i p-ANCA (mieloperossidasi). La diagnosi differenziale va posta soprattutto nei confronti di infezioni parassitarie, polmonite eosinofila, patologie mieloproliferative e sindrome ipereosinofila (HES).
Trattamento
La terapia nelle fasi acute è a base di cortisone ad alte dosi pari a 1 mg/kg/die di prednisone. In casi particolarmente aggressivi si inizia con boli di cortisone per ridurre rapidamente il livello degli eosinofili e limitare il danno tessutale e si aggiungono immunosoppressori come ciclofosfamide e methotrexate. Nelle forme ANCA positive può essere somministrato rituximab in aggiunta al cortisone. Si riduce quindi progressivamente il cortisone per arrivare al dosaggio minimo necessario per controllare la vasculite e i sintomi respiratori. Come terapia di mantenimento si possono associare metotrexato, azatioprina o micofenolato. Dal 2017 si introduce, in associazione allo steroide, mepolizumab (anticorpo monoclonale anti-IL-5) che porta ad una riduzione rapida degli eosinofili con un ottimo controllo dell’asma e della rinosinusite.
La prognosi è favorevole e le recidive, più frequenti nel primo anno dopo la diagnosi, sono rare nelle forme ben controllate dalla terapia medica e seguite regolarmente nei centri di riferimento per la EGPA. Con le nuove terapie biologiche si riducono inoltre molti effetti avversi causati dalla precedente terapia sistemica con corticosteroidi.
Conclusioni
Per questa patologia è particolarmente importante la collaborazione multidisciplinare, in quanto possono essere colpiti quasi tutti gli organi; è pertanto necessario assicurare la massima competenza e conoscenza da parte di specialisti, anche in collaborazione attiva con l’Associazione dei pazienti APACS APS (apacs-egpa.org).
Come per tutte le malattie rare è importante che i professionisti medici, specialisti e di medicina generale, siano a conoscenza dei possibili differenti quadri di presentazione della patologia, al fine di poter effettuare una corretta diagnosi. Gli specialisti potenzialmente coinvolti nella diagnosi sono infatti numerosi, tra cui: lo Pneumologo per l’asma grave con interessamento nasale e l’ipereosinofilia, l’ORL per le forme recidivanti di poliposi nasale, l’Allergo-Immunologo e il Reumatologo per l’ipereosinofilia e la vasculite, il Nefrologo per le diverse forme di glomerulonefrite, il Cardiologo per la miocardite o la trombosi cardiaca, il Neurologo per la neuropatia con eosinofilia, il Medico del Pronto Soccorso per un quadro clinico complesso associato a ipereosinofilia.
Anche il MMG è fortemente coinvolto, in quanto può osservare inizialmente il paziente e le sue successive manifestazioni cliniche.
È molto utile inoltre l’applicazione di un PDTA condiviso tra i diversi attori, per uniformare il percorso diagnostico, terapeutico e riabilitativo dei pazienti, che vede la forte collaborazione tra paziente e medico in questo percorso assistenziale a volte impegnativo.
Bibliografia
- Trivoli G, Terrier B, Vaglio A. Eosinophilic granulomatosis with polyangiitis: understanding the disease ant its management. Rheumatology 2020;59:iii84-iii94.
- Schroeder JW, Folci M, Sinico RA, et al. Anti-neutrophil cytoplasmatic antibodies posiitivity and anti-leukotrienes in eosinophilic granulomatosis with polyangiitis: A retrospective monocentric study on 134 patients. Int Arch Allergy Immunol. 2019;180:64-71.
- Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. New Engl J Med. 2017;376:1921-32.
- Bettiol A, Schroeder JW, Emmi G, et al. Mepolizumab for Eosinophilic Granulomatosis with Polyangiitis: a European multicenter observational study. Arthritis Rheumatol. 2022;74(2):295-306.
- Groh M, Pagnoux C, Baldini C, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med. 2015;26(7):545-53.
- Wechsler ME, Hellmich B, Roufosse F, et al. Unmet needs and evidence gaps in hypereosinophilic syndrome and eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol. 2023;151(6):1415-1428.
- Furuta S, Iwamoto T, Nakajima H. Update on eosinophilic granulomatosis with polyangiitis. Allergol Int. 2019;68(4):430-436.
- Emmi G, Bettiol A, Durante E, et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol. 2023;19(6):378-393.