




la Newsletter

Fiorina Giona, Simona Bianchi
Dipartimento di Medicina Traslazionale e di Precisione - Sapienza Università di Roma
La malattia di Rosai-Dorfman: una rara tra le rare | Sebbene la...
Sebbene la RDD presenti caratteristiche peculiari, la diagnosi...
 Nel novembre 2013, ad un uomo di 63 anni con adenomegalie laterocervicali e sottomandibolari bilaterali veniva posta diagnosi di malattia di Rosai-Dorfman (RDD), S100+, CD68KP1+, CD68RPGM1+, CD1a-, langherina (CD207)-, CKAE1AE3-, CD30-, ALKp80-, MPO-, CD34-, CD117-. Il paziente iniziava terapia steroidea al dosaggio di 0,5 mg/Kg/die, con miglioramento del quadro clinico. Per ricomparsa di adenomegalie diffuse e localizzazione di malattia a livello dei seni paranasali, giungeva alla nostra osservazione nell’ottobre 2015.
Nel novembre 2013, ad un uomo di 63 anni con adenomegalie laterocervicali e sottomandibolari bilaterali veniva posta diagnosi di malattia di Rosai-Dorfman (RDD), S100+, CD68KP1+, CD68RPGM1+, CD1a-, langherina (CD207)-, CKAE1AE3-, CD30-, ALKp80-, MPO-, CD34-, CD117-. Il paziente iniziava terapia steroidea al dosaggio di 0,5 mg/Kg/die, con miglioramento del quadro clinico. Per ricomparsa di adenomegalie diffuse e localizzazione di malattia a livello dei seni paranasali, giungeva alla nostra osservazione nell’ottobre 2015.
Iniziava trattamento con interferon alfa-2° 3.000.000 UI/die x 5 giorni a settimana, con scomparsa della malattia nelle sedi affette. A dicembre 2019, per una recidiva della RDD, veniva trattato con terapia steroidea al dosaggio di 0,5 mg/Kg/die, con scarsa risposta, per cui a ottobre 2020 iniziava trattamento con cicli di ciclofosfamide 100 mg/die per os x 10 giorni ogni 28 giorni. Dopo 5 cicli, per persistenza delle linfoadenomegalie, si iniziava terapia con interferon alfa-2b 180 mcg/settimana con scomparsa dell’obiettività clinica. Il paziente è in ottime condizioni generali e ha sempre avuto una buona qualità di vita.
La malattia di Rosai-Dorfman
La RDD ha una prevalenza di 1/200.000. Colpisce prevalentemente giovani adulti con leggera predominanza per il sesso maschile e la razza africana (1). L’eziopatogenesi della RDD è sconosciuta. Grazie alle nuove scoperte sulle basi molecolari della malattia, la RDD è stata riconosciuta come un processo mieloproliferativo con mutazioni in ARAF, MAP2K1, NRAS e KRAS nella RDD nodale ed extranodale, ma non cutanea.
La RDD classica nodale si presenta con una massiccia linfoadenopatia cervicale bilaterale indolore, spesso con sintomi (febbre associata, perdita di peso e sudorazione notturna). Possono essere coinvolti anche i linfonodi inguinali, retroperitoneali e mediastinici. Le localizzazioni extranodali, riportate in oltre il 40% dei casi, coinvolgono prevalentemente cute, cavità nasale, osso, tessuto orbitale e sistema nervoso centrale, prevalentemente durale.
Diagnosi
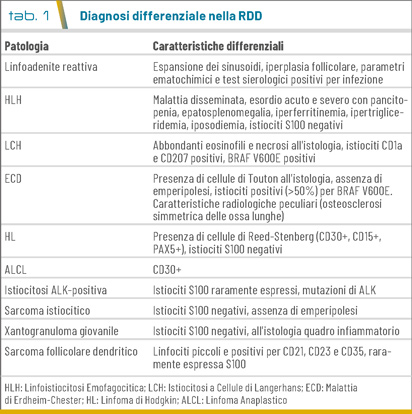
 Sebbene la RDD presenti molte caratteristiche peculiari, la diagnosi differenziale tra RDD e altri disordini istiocitari è talvolta difficile (Tab. 1).
Sebbene la RDD presenti molte caratteristiche peculiari, la diagnosi differenziale tra RDD e altri disordini istiocitari è talvolta difficile (Tab. 1).
La diagnosi di certezza si basa sul riscontro del fenomeno dell’emperipolesi, cioè la penetrazione attiva in un istiocita di una cellula, perlopiù linfocitaria, che rimane intatta al suo interno (Fig. 1) (2), e sulle caratteristiche immunocitochimiche (3). Le cellule RDD sono macrofagi CD14+, HLA-DR+, CD68++, CD163+, S100+ e fascina+, tipicamente negative per CD1a e CD207.
La prognosi è correlata al numero di stazioni linfonodali e/o alle sedi extranodali coinvolte, ed è generalmente peggiore in presenza di una malattia immunologica associata (4,5).
Trattamento
Una remissione spontanea è possibile nel 50% dei casi di RDD isolata. Per via dell’alto tasso di remissione spontanea, il watch and wait è la scelta più ragionevole in molti casi di RDD (6).
Quando la malattia è sintomatica ed è necessario un debulking, possono essere utilizzate diverse opzioni terapeutiche, che vanno dalla chirurgia, alla chemioterapia (mono o polichemio), agli interferoni e alla radioterapia, fino ai farmaci target (Tab. 2).
La durata ottimale delle diverse terapie non è nota: l’approccio più ragionevole è eseguire da 6 a 12 mesi di terapia sistemica seguita dall’osservazione clinica.

Una prima valutazione della risposta deve essere effettuata entro 4 mesi dallo stop terapia; se la malattia si stabilizza o è in remissione, l’intervallo di sorveglianza può essere poi esteso a 6-12 mesi (7). Circa il 10% dei pazienti può sviluppare complicanze severe, quali infezioni e amiloidosi (8,9).
Considerata la rarità della patologia non è ancora definito il percorso terapeutico più appropriato: trial clinici prospettici, unitamente alla scoperta di nuove mutazioni, potrebbero, nel prossimo futuro, portare ad un approccio terapeutico personalizzato.
Bibliografia
- Galicier L, Fieschi C, Meignin V, et al. Rosai-Dorfman disease. Presse Med. 2007;36(11):1669-75.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61(7):1329-35.
- Paulli M, Rosso R, Kindl S, et al. Immunophenotypic characterization of the cell infiltrate in five cases of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Hum Pathol. 1992;23(6):647-54.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman Disease between Proliferation and Neoplasia. Cancers (Basel). 2022;14(21):5271.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7(1):19-73.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73(11):697-705.
- Younes IE, Sokol L, Zang L. Rosai–Dorfman Disease between Proliferation and Neoplasia. Cancers (Basel). 2022; 14(21): 5271.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J ClinPathol. 2020;73(11):697-705.
- Lima FB, Barcelos PS, Constâncio AP, et al. Rosai-Dorfman disease with spontaneous resolution: case report of a child. Rev Bras Hematol Hemoter. 2011;33(4):312-4.