




la Newsletter

Alessia Carrer1,2,Milena Mariani2
1Università degli Studi di Milano - Scuola di Specializzazione in Genetica Medica; 2UOC Pediatria. Presidio S. Fermo. ASST-Lariana, Como. Centro Fondazione Mariani per il Bambino Fragile
La sindrome KBG | La sindrome KBG è un disordine del...
La sindrome KBG è un disordine del neurosviluppo causato da...
M. è una ragazza di 13 anni, giunta in visita presso il nostro ambulatorio di Genetica Pediatrica. I genitori riferiscono che la gravidanza e il periodo neonatale di M. sono decorsi regolarmente, così come il raggiungimento delle prime tappe di sviluppo psicomotorio (deambulazione autonoma a 15 mesi, prime parole a 12 mesi). I primi segnali di preoccupazione sono stati un lento accrescimento staturo-ponderale, accompagnato da scarso appetito e frequenti rigurgiti, e alcune difficoltà scolastiche manifestate all’inizio della scuola primaria, per cui M. ha intrapreso percorsi di psicomotricità, logopedia e pedagogia. Ora M. presenta una statura ai limiti inferiori della norma, con altezza al 10° percentile, peso al 3°-10° percentile e circonferenza cranica al 3°-10° percentile; i test di sviluppo (scala WISC IV) mostrano uno sviluppo cognitivo al limite di norma (QIT 89) e le principali difficoltà riguardano l’area comportamentale/relazionale, con alcuni episodi di agitazione psicomotoria.
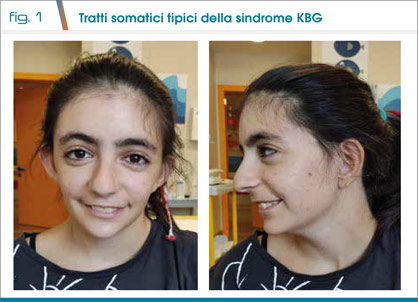 I peculiari tratti del volto hanno suggerito l’ipotesi diagnostica “a colpo d’occhio”: volto triangolare, attaccatura bassa dei capelli, sopracciglia folte con accenno a sinofria, ciglia lunghe, punta nasale bulbosa, labbro superiore sottile con commissure labiali rivolte verso il basso, palato ogivale, padiglioni auricolari lievemente anteversi (Fig. 1); fondamentale per il sospetto clinico di sindrome KBG è stata la “maniglia diagnostica” costituita dagli incisivi centrali larghi e a margini frastagliati.
I peculiari tratti del volto hanno suggerito l’ipotesi diagnostica “a colpo d’occhio”: volto triangolare, attaccatura bassa dei capelli, sopracciglia folte con accenno a sinofria, ciglia lunghe, punta nasale bulbosa, labbro superiore sottile con commissure labiali rivolte verso il basso, palato ogivale, padiglioni auricolari lievemente anteversi (Fig. 1); fondamentale per il sospetto clinico di sindrome KBG è stata la “maniglia diagnostica” costituita dagli incisivi centrali larghi e a margini frastagliati.
M. presenta anche alcune sfumate peculiarità scheletriche: abbozzo di costa cervicale accessoria, assottigliamento delle falangi ungueali e clinodattilia del V dito della mano.
Lo screening malformativo (ecografia addominale, ecocardiografia, RMN encefalo) ha evidenziato unicamente la presenza di alcuni dismorfismi cerebrali aspecifici (aspetto tozzo del corpo calloso, ipoplasia vermiana) e noduli di eterotopia emisferica cerebellare.
L’analisi del gene ANKRD11 ha identificato una variante patogenetica di origine de novo, confermando la diagnosi di sindrome KBG.
La sindrome KBG
La sindrome KBG è una condizione genetica descritta per la prima volta nel 1975, il cui nome deriva dalle iniziali dalle prime tre famiglie in cui è stata osservata.
È un disordine del neurosviluppo causato da aploinsufficienza del gene ANKRD11, che, oltre all’impatto sullo sviluppo cognitivo, può comportare complicanze in diversi organi e apparati.
La macrodontia degli incisivi centrali superiori è il principale elemento di sospetto clinico, che può suggerire la diagnosi “a colpo d’occhio”, soprattutto quando si presenta in associazione a facies caratteristica, ritardo di sviluppo psicomotorio/disabilità intellettiva, problematiche comportamentali e bassa statura.
La sindrome KBG è una condizione rara, ma probabilmente sottodiagnosticata per la possibile presenza di caratteristiche solo sfumate e aspecifiche. Ad oggi in letteratura sono stati descritti circa 200 individui affetti.
Elementi clinici di sospetto diagnostico
È molto difficile sospettare la sindrome KBG prima dell’eruzione dei denti permanenti.
Nell’inquadramento diagnostico di un bambino con ritardo psicomotorio, il sospetto clinico di sindrome KBG si basa principalmente sulla presenza di una facies suggestiva. Gli elementi dismorfologici caratteristici, in parte osservabili anche nel caso clinico presentato, sono brachicefalia, volto triangolare, impianto basso anteriore dei capelli, sinofria con sopracciglia larghe e folte, occhi spaziati, radice nasale prominente, punta nasale bulbosa, narici anteverse, filtro lungo, labbro superiore sottile, macrodontia (soprattutto degli incisivi centrali superiori permanenti) e orecchie prominenti.
 Ad oggi non ci sono dei criteri per una diagnosi clinica, ma diversi autori hanno suggerito possibili elementi per il sospetto diagnostico (Tab. 1). La diagnosi viene confermata dall’esito positivo del test genetico.
Ad oggi non ci sono dei criteri per una diagnosi clinica, ma diversi autori hanno suggerito possibili elementi per il sospetto diagnostico (Tab. 1). La diagnosi viene confermata dall’esito positivo del test genetico.
Dati auxologici
I parametri alla nascita sono generalmente normali, tuttavia si collocano frequentemente nei percentili inferiori al range di normalità. La curva di crescita tende a deflettersi intorno ai 10 anni di età e la bassa statura si osserva nel 40-77% dei casi.
È possibile il riscontro di ritardo nella maturazione ossea, mentre in genere la valutazione endocrinologica non rileva anomalie ormonali.
Sviluppo psico-intellettivo e fenotipo comportamentale
L’impatto della sindrome KBG sulle capacità cognitive è molto variabile. Più del 90% dei bambini manifesta un certo grado di ritardo di sviluppo psicomotorio, soprattutto del linguaggio. Non è riportata in letteratura la regressione delle capacità acquisite.
Solo una minoranza di persone adulte non ha difficoltà cognitive, mentre la maggioranza (70%) presenta disabilità intellettiva di grado lieve; più rara è una compromissione più severa. Più della metà degli adulti ha un’attività lavorativa ed è autosufficiente nelle attività quotidiane.
Una problematica rilevante, manifestata da più della metà degli affetti, è la presenza di difficoltà di concentrazione, timidezza, disturbo d’ansia e aggressività.
Malformazioni maggiori e complicanze mediche associate
Dal punto di vista malformativo, l’apparato scheletrico è maggiormente coinvolto: sono frequenti anomalie costovertebrali (coste cervicali, vertebre dismorfiche), scoliosi, anomalie del palato e dello sterno, brachidattilia/clinodattilia e ritardo di chiusura della fontanella anteriore. Più rara è la presenza di difetti cardiaci (10-15%).
La Risonanza Magnetica dell’encefalo rivela anomalie in quasi la metà dei casi, con possibile riscontro di ipoplasia del verme cerebellare, allargamento della cisterna magna, cisti aracnoidee e ipoplasia del nervo ottico.
In circa la metà dei soggetti sono presenti anche anomalie all’elettroencefalogramma, con o senza manifestazioni epilettiche.
Nei maschi non è insolita la presenza di criptorchidismo (25-30%).
Per quanto riguarda le complicanze mediche, i genitori riportano frequentemente difficoltà di alimentazione/problematiche gastro-intestinali (vomito, stipsi, reflusso gastroesofageo), soprattutto nell’infanzia.
L’ipoacusia di grado variabile è descritta nel 25-30% dei soggetti, di tipo sia neurosensoriale che percettivo, in particolare da otite media ricorrente.
Difetto genetico di base
La sindrome KBG è compresa nel gruppo delle cromatinopatie, condizioni ereditarie causate da alterazione dei meccanismi di regolazione epigenetica della cromatina.
È causata dalla perdita di funzione del gene ANKRD11, che modula l’acetilazione degli istoni. Si tratta di una condizione ad ereditarietà autosomica dominante, per cui è sufficiente che una sola delle due copie del gene sia alterata per avere manifestazioni cliniche.
La perdita di funzione del gene ANKRD11 può essere causata sia da mutazioni puntiformi (70% dei casi) sia da delezioni nella regione 16q24.3 comprendenti il gene ANKRD11 (30% dei casi).
Non è nota una correlazione genotipo/fenotipo, ad eccezione dei casi con ampie delezioni, che coinvolgono oltre a ANKRD11 anche diversi geni limitrofi, in cui il quadro clinico è tendenzialmente più severo.
Sebbene le nuove tecniche consentano di identificare il difetto genetico in una quota sempre maggiore di soggetti con sospetto clinico di sindrome KBG, alcuni casi rimangono ancora non diagnosticati, suggerendo la presenza di ulteriori meccanismi biologici sottostanti non ancora scoperti.
Diagnosi differenziale
La principale condizione in diagnosi differenziale è la sindrome di Cornelia de Lange, per la sovrapposizione di alcuni tratti dismorfici (impianto basso anteriore dei capelli, sopracciglia folte e arcuate, labbra sottili con commissure labiali rivolte verso il basso, mento piccolo, brachidattilia). Comuni alle due condizioni sono anche lo scarso accrescimento staturo-ponderale e la possibile associazione con ipoacusia ed altre anomalie congenite (anomalie del palato, cardiopatia, criptorchidismo); più tipiche della sindrome di Cornelia de Lange sono invece la microcefalia e l’impatto più severo sulle capacità cognitive.
L’analisi genetica non è sempre dirimente tra le due condizioni, in quanto anche in soggetti con diagnosi clinica di Cornelia de Lange sono state identificate varianti patogenetiche nel gene ANKRD11, suggerendo che la sovrapposizione fenotipica rifletta una vicina base biologica tra le due sindromi.
Trattamento
I bambini e gli adulti affetti da Sindrome KBG necessitano di una presa in carico multidisciplinare, volta al potenziamento delle capacità cognitive/sociali in caso di difficoltà psicomotorie e comportamentali, al monitoraggio clinico e alla prevenzione/trattamento delle possibili complicanze mediche associate.
Evidenze preliminari suggeriscono che la terapia con ormone della crescita possa avere benefici sulla bassa statura.
Bibliografia
- Morel Swols D, Tekin M. KBG Syndrome. 2018 Mar 22. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2022.
- Loberti L, et al. Natural history of KBG syndrome in a large European cohort. Hum Mol Genet. 2022 Jul 21:ddac167. doi: 10.1093/hmg/ddac167. Epub ahead of print. PMID: 35861666
- Bestetti I, et al. Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome. Int J Mol Sci. 2022;23(11):5912.
- Low K, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet Part A. 2016; 170(11):2835-2846.