




la Newsletter

Barbara Ruggiero, Francesco Emma
UOC di Nefrologia, Ospedale Pediatrico Bambino Gesù, IRCCS, Roma
La sindrome di Fanconi | Causata da una disfunzione delle cellule...
Causata da una disfunzione delle cellule tubulari prossimali del...
La sindrome di Fanconi è causata da una disfunzione globale delle cellule tubulari prossimali del rene. Questa porzione del nefrone è metabolicamente molto attiva ed ha la funzione di riassorbire la maggior parte dell’acqua, dei sali e di altre sostanze filtrate dai glomeruli. Ne consegue una perdita massiva nelle urine di acqua e soluti, in particolare di sali, aminoacidi, glucosio, fosfati, bicarbonati, e proteine di basso peso molecolare.
Epidemiologia
La sindrome di Fanconi in età pediatrica ha generalmente una causa genetica. L’incidenza esatta non è nota. La cistinosi, causa principale di sindrome di Fanconi nella prima infanzia, ha un’incidenza di 1:150.000.
Manifestazioni cliniche
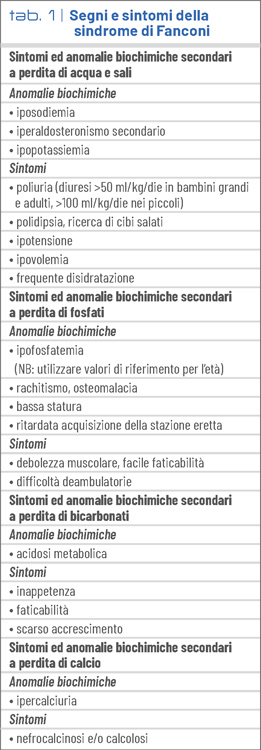 L’entità della disfunzione tubulare prossimale è variabile. L’elemento costante è la presenza nelle urine di proteine di basso peso molecolare. I sintomi sono altrettanto variabili e dipendono dalla precocità e dalla severità del danno tubulare (Tab. 1). Le perdite urinarie di glucosio, amino acidi e proteine di basso peso molecolare in genere non causano sintomi particolari.
L’entità della disfunzione tubulare prossimale è variabile. L’elemento costante è la presenza nelle urine di proteine di basso peso molecolare. I sintomi sono altrettanto variabili e dipendono dalla precocità e dalla severità del danno tubulare (Tab. 1). Le perdite urinarie di glucosio, amino acidi e proteine di basso peso molecolare in genere non causano sintomi particolari.
I pazienti possono sviluppare segni evidenti di sotto idratazione. L'ipopotassiemia è un fenomeno comune, derivante sia dalle perdite urinarie di potassio che dall'aumento dell'attività dell'aldosterone secondario alla condizione di ipovolemia cronica. L'acidosi metabolica è spesso molto marcata e difficile da correggere. Quando è presente, l'ipofosfatemia può causare lesioni di rachitismo ed osteomalacia.
Nelle forme ad esordio molto precoce i pazienti sviluppano generalmente un ritardo di crescita secondario a rachitismo, carenza calorica, acidosi, inappetenza, ipovolemia cronica. L’ipercalciuria non determina ipocalcemia, ma può causare nefrocalcinosi e/o nefrolitiasi.
Eziologia e patogenesi
La sindrome di Fanconi può essere causata da fattori genetici o acquisiti, con quest'ultima condizione spesso derivante dall'azione lesiva di agenti tossici a livello tubulare. La sindrome di Fanconi viene classificata come idiopatica quando non è possibile identificarne la causa specifica.
La sequenza di eventi che conducono alla disfunzione globale della cellula tubulare prossimale è variabile a seconda della sua eziologia. Nelle forme genetiche, i difetti possono interessare i processi di traffiking intracellulare, in particolare quelli legati all'endocitosi mediata dai recettori cubilina e megalina, il metabolismo intermedio, il metabolismo energetico, o compromettere la funzione di trasportatori basolaterali. Ne risulta un difetto globale del funzionamento della cellula con carenza energetica.
Trattamento
Fatta eccezione per la cistinosi, non esiste una terapia eziologica per la maggior parte delle forme di sindrome di Fanconi. La terapia è sostanzialmente sintomatica, volta alla correzione degli squilibri elettrolitici, dell'acidosi, e del rachitismo. In funzione dei sintomi manifestati, la terapia può includere supplementi di bicarbonato di sodio, citrato di potassio, sodio e potassio cloruro, fosfati e/o vitamina D. L’ipovolemia richiede un adeguato apporto di liquidi, con particolare attenzione alle condizioni che possono favorire una disidratazione. Nei bambini più piccoli può essere necessario usare il sondino nasogastrico ed in alcuni casi una gastrostomia. Nelle forme secondarie, il trattamento comprende la terapia della patologia di base, ove possibile.
Forme genetiche
La cistinosi è la prima causa di sindrome di Fanconi nei bambini in età prescolare (Tab. 2). È una malattia sistemica, dovuta a varianti patogenetiche del gene CTNS che codifica per la cistinosina, un trasportatore lisosomiale di cistina, con conseguente accumulo intralisosomiale di cistina in vari organi e tessuti. Sono coinvolti rene, osso, occhio, ghiandole endocrine (con sviluppo di diabete, ipotiroidismo, ipogonadismo), cute, muscolo ed encefalo. La forma infantile è la più grave e anche la più frequente. I bambini manifestano generalmente i primi sintomi legati alla sindrome di Fanconi intorno ai 4-6 mesi di vita. La diagnosi si basa sull’analisi genetica e sul dosaggio della cistina intraleucocitaria. Cristalli di cistina sono obiettivabili nell’occhio, tuttavia la loro assenza non esclude la malattia nei primi due anni di vita. La terapia comprende la somministrazione di cisteamina che consente di ridurre l’accumulo di cistina nelle cellule.
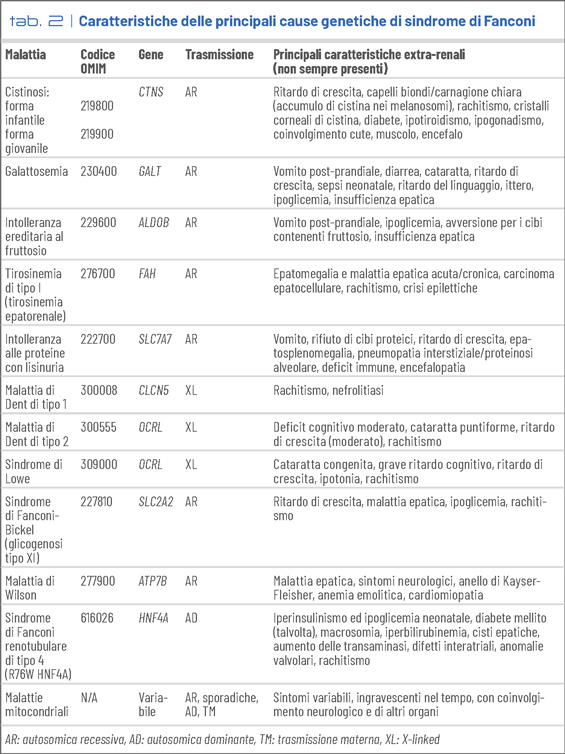
La sindrome di Dent è una tubulopatia legata al cromosoma X, di cui si riconoscono due forme: la malattia di Dent di tipo 1, dovuta a mutazioni del gene che codifica per il canale del cloro CLC-5, e la malattia di Dent di tipo 2 che, insieme alla sindrome oculo-cerebro-renale di Lowe, deriva da mutazioni del gene OCRL. È sempre presente una proteinuria di basso peso molecolare. La seconda anomalia più frequente, riscontrata in >80% dei pazienti, è l’ipercalciuria. L’evoluzione è verso l’insufficienza renale terminale generalmente nella terza e quarta decade di vita. I pazienti con sindrome di Dent di tipo 2 possono avere inoltre anche lievi deficit cognitivi, bassa statura e cataratta puntiforme. La sindrome di Lowe ha un coinvolgimento molto più severo, con cataratta congenita, grave deficit cognitivo, ipotonia generalizzata e disfunzione tubulare più marcata.
Il citrato di potassio può essere utile per contrastare l’ipercalciuria nella sindrome di Dent. In modelli murini, la supplementazione con citrato di potassio ha anche un effetto protettivo sulla progressione dell’insufficienza renale.
Tra i disordini del metabolismo dei carboidrati, vi sono la galattosemia, dovuta alla carenza dell’enzima degradante il galattosio e l’intolleranza ereditaria al fruttosio, causata dal deficit dell’enzima degradante il fruttosio. In genere la diagnosi viene fatta in base ai sintomi extrarenali.
La sindrome di Fanconi-Bickel, è dovuta a mutazioni del trasportatore basolaterale di glucosio GLUT2 che media il trasporto del glucosio sia nel tubulo prossimale che a livello del fegato. Il difetto causa un accumulo di glicogeno a livello renale ed epatico. I pazienti presentano, oltre alla sindrome di Fanconi, anche ipoglicemia a digiuno.
La sindrome di Fanconi renotubulare di tipo 4 con diabete giovanile ad esordio in età adulta è causata da una variante patogenetica particolare (R76W) del gene HNF4A. I pazienti possono manifestare iperinsulinismo neonatale ed ipoglicemia, bassa statura, anomalie epatiche e cardiache, e diabete mellito in età giovane-adulta.
Tra i disordini del metabolismo proteico, va ricordata la tirosinemia ereditaria di tipo 1 causata dal deficit di attività dell’enzima fumaril-acetoacetato idrolasi, con conseguente formazione di succinil-acetone che danneggia il metabolismo delle cellule tubulari prossimali renali. La terapia si basa su una dieta a basso contenuto di fenilalanina e tirosina e sull’uso del nitisinone che blocca la formazione di succinil-acetone ed attenua significativamente il danno tubulare prossimale.
Tra i disordini del metabolismo dei metalli, va menzionato il morbo di Wilson, causato da un difetto del trasporto del rame. Raramente, tuttavia, i primi sintomi sono legati alla sindrome di Fanconi. La terapia con penicillamina, se instituita precocemente, può far regredire le manifestazioni cliniche.
Infine, molte mitocondriopatie possono causare delle sindromi di Fanconi più o meno severe, nell’ambito di quadri clinici solitamente molto gravi con coinvolgimento sistemico (il sistema nervoso è sempre coinvolto). Non esistono terapie specifiche, salvo nei difetti di biosintesi del coenzima Q10. Trattamenti con menadione, ubidecarenone, riboflavina, o acido ascorbico sono talvolta utilizzati con risultati tuttavia modesti.
Diagnosi differenziale
Esistono difetti selettivi del riassorbimento di soluti o delle proteine di basso peso molecolare a livello prossimale che non configurano un quadro di sindrome di Fanconi. Trattasi in genere di difetti di trasporto a livello della membrana apicale che possono causare perdite anormali di glucosio, aminoacidi, fosforo o bicarbonato. In questi casi, la proteinuria di basso peso molecolare è sempre assente o molto modesta.
L’acidosi tubulare distale può mimare all’esordio una sindrome di Fanconi con disfunzione generalizzata del tubulo prossimale e del tubulo distale; possono essere presenti aminoaciduria, uricosuria, perdita di sodio, che regrediscono non appena l’acidosi metabolica viene corretta dalla terapia.
Raramente i pazienti possono presentare una proteinuria di basso peso molecolare secondaria a varianti patogenetiche dei geni che codificano per i recettori cubilina/amnionless o megalina responsabili dell’endocitosi mediata da recettori. In questo caso, la proteinuria è isolata.
Le varianti che interessano il complesso cubilina/amnionless causano la sindrome di Imerslund-Gräsbeck che si caratterizza anche da anemia megaloblastica, essendo queste proteine responsabili anche del riassorbimento intestinale di vitamina B12, mentre le alterazioni di megalina causano la sindrome di Donnai-Barrow, estremamente rara e caratterizzata da un grave quadro malformativo.
Forme secondarie o acquisite
Numerose sostanze possono danneggiare il tubulo prossimale renale. La gravità e la reversibilità del danno tubulare varia in base al tipo di tossina, alle caratteristiche di esposizione (durata, entità) e alle caratteristiche intrinseche del paziente. Un tempo, una delle maggiori cause di sindrome di Fanconi era l’intossicazione da metalli pesanti tra cui il piombo, che si associa spesso ad insufficienza renale cronica e danni al sistema nervoso, ed il cadmio, che è particolarmente tossico a livello osseo.
Tali intossicazioni sono ormai rare nei paesi occidentali; quando si verificano, sono generalmente secondarie a contaminazioni industriali del suolo.
Oggigiorno, i farmaci chemioterapici rappresentano una causa importante di sindrome di Fanconi acquisita; in particolare cisplatino e ifosfamide, la cui tossicità tubulare è dose-dipendente e spesso irreversibile. Gli altri farmaci maggiormente implicati nella sindrome di Fanconi sono gli antivirali, ad esempio il cidofovir, gli antibiotici aminoglicosidi, e gli antiepilettici, tra cui soprattutto l’acido valproico.
Alcune malattie sistemiche possono secondariamente causare una sindrome di Fanconi. Tra esse va menzionato il mieloma multiplo e le paraproteinemie che possono causare deposizione e cristallizzazione di catene leggere a livello del tubulo prossimale.
Malattie autoimmuni quali la sindrome di Sjögren o il lupus eritematoso sistemico possono causare un danno tubulare, generalmente diffuso o distale, ma talvolta limitato alle cellule del tubulo prossimale.
Bibliografia
- Sirac C, Bridoux F, Essig M, et al. Toward understanding renal Fanconi syndrome: step by step advances through experimental models. Contrib Nephrol. 2011; 169: 247-61.
- Foreman JW. Fanconi Syndrome. Pediatr Clin North Am. 2019; 66(1):159-167.
- Hoogstraten CA, Hoenderop JG, de Baaij JHF. Mitochondrial Dysfunction in Kidney Tubulopathies. Annu Rev Physiol. 2024; 86: 379-403.
- Devuyst O, Thakker RV. Dent's disease. Orphanet J Rare Dis. 2010; 5: 28.
- Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. 2014; 29 Suppl 4(Suppl 4): iv87-94.
- Igarashi T, Emma F, and Hayes W. Fanconi syndrome. In Pediatric Nephrology 8th edition, Emma F, Goldstein S, Bagga A, Bates CM and Shroff R eds, Springer Nature Switzerland AG, 849-876, 2022.