




la Newsletter

Daniele Vitale, Elisabetta Prada
UOC Pediatria - Centro Fondazione Mariani per il Bambino Fragile, ASST-Lariana, Como
La sindrome di Waardenburg | Caratterizzata da ipoacusia...
Caratterizzata da ipoacusia neurosensoriale e disturbi della...
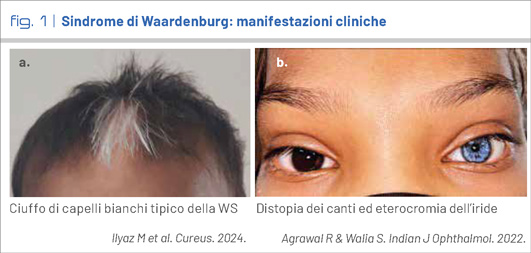
A., 9 anni, secondogenita di genitori sani non consanguinei, nasce a 37 settimane da parto eutocico con buon adattamento alla vita extrauterina e parametri auxologici nella norma. Ai due mesi di vita è diagnosticata una ipoacusia neurosensoriale bilaterale di grado severo, per cui la bimba si sottopone ad impianto cocleare. Sin dai primi anni di vita, la madre riferisce per A. una stipsi marcata; approfondimenti strumentali e istologici successivi caratterizzano un quadro di agangliosi colica (malattia di Hirschsprung). L’osservazione clinica chiarisce ogni dubbio: un ciuffo di capelli bianchi frontale e l’eterocromia dell’iride ci indicano una diagnosi ben precisa.
La sindrome di Waardenburg
Condizione genetica caratterizzata da ipoacusia neurosensoriale e disturbi della pigmentazione di cute, capelli o iride, la sindrome di Waardenburg (WS) risulta estremamente variabile in termini di espressione clinica e severità globale, anche all’interno della stessa famiglia. L’ipoacusia neurosensoriale rappresenta uno degli aspetti clinici più comuni nei pazienti affetti da WS (70% circa) ed è tipicamente congenita e di grado profondo.
Le anomalie della pigmentazione sono presenti in una percentuale variabile tra il 30 ed il 55% e includono la presenza di un ciuffo bianco di capelli tipicamente frontale (Fig. 1 a), presente alla nascita o a comparsa entro l’adolescenza, l’incanutimento precoce dei capelli, eterocromia iridea completa o parziale o più raramente iridi ipoplasiche di colore blu brillante, discromie cutanee come macule ipo- o iper-pigmentate. La caratteristica facciale più tipica è la distopia dei canti, o telecanto (Fig. 1 b), ossia un aumento della distanza tra i canti mediali degli occhi con una distanza interpupillare normale, valutato mediante l’indice W, parametro biometrico calcolato sulla base della misura delle distanze intercantale interna, interpupillare e intercantale esterna. L’incidenza stimata è pari a circa 1:40.000; considerata l’ampia variabilità di espressione e la possibilità di forme con presentazione molto sfumata è possibile che il dato sia sottostimato.
Criteri diagnostici
La WS è ulteriormente classificata in quattro sottotipi, sulla base della presentazione clinica.
I criteri clinici diagnostici sono stati stabiliti dal Consorzio della WS nel 1992:
- Criteri maggiori: sordità neurosensoriale congenita; ciocca ipocromica frontale; alterazioni della pigmentazione dell’iride; distopia dei canti (indice W >1,95); familiare di primo grado affetto.
- Criteri minori: aree ipocromiche cutanee; sinofria o slargamento delle sopracciglia nella parte centrale; naso con radice alta/ampia; ipoplasia delle ali nasali; incanutimento precoce dei capelli.
La diagnosi clinica di WS è raggiunta in caso di presenza di 2 criteri maggiori oppure di 1 criterio maggiore e di 2 minori. Se la distopia dei canti rappresenta uno dei criteri soddisfatti, si pone diagnosi di WS tipo 1; in caso contrario si pone diagnosi di WS tipo 2. La diagnosi di WS tipo 3 è formulata in caso di criteri clinici soddisfatti per WS in associazione ad anomalie muscolo-scheletriche. La diagnosi di WS tipo 4 è formulata in caso di criteri clinici soddisfatti per WS in associazione a malformazione di Hirschsprung.
Crescita e sviluppo
L’accrescimento staturo-ponderale risulta nella norma, ad eccezione di pazienti con WS di tipo 4, in cui la crescita può risultare rallentata per la presenza di complicanze gastro-intestinali. Dal punto di vista psicomotorio, lo sviluppo è generalmente nella norma; il deficit uditivo può associarsi ad un ritardo nell’acquisizione del linguaggio.
Malformazioni e complicanze
I soggetti con WS tipo 3 presentano un coinvolgimento dell’apparato muscolo-scheletrico degli arti superiori con ipoplasia muscolare, contratture in flessione dei gomiti, anomalie del carpo o sindattilie delle mani. Il quadro clinico di soggetti con WS tipo 4 è caratterizzato dalla presenza di megacolon agangliare congenito; più raramente sono descritte malformazioni ano-rettali, atresia esofagea. Nella WS tipo 4, in particolare in forme SOX10-correlate, può essere inoltre presente un coinvolgimento neurologico con demielinizzazione centrale e periferica.
Prognosi
La prognosi è generalmente favorevole, con una normale aspettativa di vita. Per i pazienti con diagnosi di WS tipo 4 la prognosi è influenzata dalla presenza di eventuali complicanze gastro-intestinali.
Counseling genetico
La sindrome di Waardenburg è generalmente trasmessa con modalità autosomica dominante (AD) ed è spesso ereditata da un genitore affetto (in alcuni casi paucisintomatico); più rare le forme a trasmissione autosomica recessiva (AR).
Si tratta di una condizione geneticamente eterogenea, le cui basi molecolari non sono tuttora pienamente comprese. Sono ad oggi noti sei geni associati alla sindrome: PAX3 (associato a WS di tipo 1 AD e a WS di tipo 3 AD o AR), MITF e SNAI2 (associati a WS di tipo 2 AD), EDNRB e EDN3 (associati a WS di tipo 4 AD o AR), e SOX10 (associato a WS di tipo 2 AD o WS di tipo 4 AD).
Bibliografia
- Fisch L. Deafness as part of an hereditary syndrome. J Laryngol Otol 1959;73:355-82.
- Farrer LA, Grundfast KM, Amos J, et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS consortium. Am J Hum Genet. 1992;50(5):902-13.
- Pingault V, Ente D, Dastot-Le Moal F, et al. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391-406.
- Milunsky JM. Waardenburg Syndrome Type I. 2001 Jul 30 [updated 2022 Oct 20]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025.
- Huang S, Song J, He C, et al. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 2022;29(9):479-497.
- Kankipati SM, Mahalingam A, Reshie A, et al. Clinical Insights Into Waardenburg-Shah Syndrome: A Case Series and Literature Review. Cureus. 2024;16(5):e59858.
- Song J, Feng Y, Acke FR, et al. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet. 2016;89(4):416-425.