




la Newsletter

Lorenzo Maggi1, Luisa Politano2
1Unità Operativa di Neuroimmunologia e Malattie Neuromuscolari, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano; 2Già Professore di Genetica Medica - Università della Campania Luigi Vanvitelli, Presidente dell'Associazione Centro Gaetano Torre per le Malattie Muscolari, Marano di Napoli
Le laminopatie muscolari scheletriche | Rientrano nel gruppo delle...
Rientrano nel gruppo delle laminopatie muscolari scheletriche la...
La lamina A/C è una proteina coinvolta in numerosi processi cellulari, pertanto mutazioni nel rispettivo gene (LMNA) sono associate ad un'ampia gamma di quadri clinici, da patologie muscolari, cardiache e metaboliche a sindromi da invecchiamento precoce (1).
Tuttavia, le malattie caratterizzate da interessamento muscolare cardiaco e scheletrico sono le più frequenti (1).
Inquadramento
Rientrano nel gruppo delle laminopatie muscolari scheletriche:
- la Distrofia Muscolare di Emery-Dreifuss autosomica dominante (EDMD2, OMIM#181350);
- la Distrofia Muscolare dei Cingoli di tipo 1B (LGMD1B, OMIM#150330 159001);
- una forma di distrofia musco-lare congenita (L-CMD, OMIM #613205),
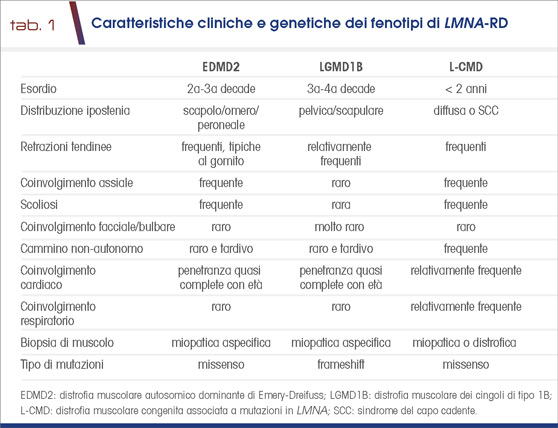
tutte causate da mutazioni nel gene LMNA e potenzialmente coesistenti nella stessa famiglia (2). Per tale motivo, e tenuto conto della considerevole sovrapposizione clinica, questi fenotipi possono essere considerati come un continuum nello spettro clinico delle distrofie da mutazioni nel gene LMNA (LMNA-RD) (2). Il cuore, essendo un muscolo, può essere coinvolto in tutti i tipi su menzionati, precedendo talvolta la comparsa della debolezza muscolare. Non esistono dati epidemiologici certi inerenti queste miopatie, che sono inserite tra le patologie rare. Le principali caratteristiche dei tre fenotipi clinici di LMNA-RD sono riportate in tabella 1.
 La EDMD2 è stato il primo fenotipo da mutazioni in LMNA descritto in letteratura, ed è caratterizzato dalla seguente triade clinica:
La EDMD2 è stato il primo fenotipo da mutazioni in LMNA descritto in letteratura, ed è caratterizzato dalla seguente triade clinica:
- precoci retrazioni tendinee a livello di caviglia, gomito e colonna;
- ipotrofia e ipostenia muscolare, a prevalente distribuzione scapolo-omero-peroneale, in particolare nelle prime fasi di malattia;
- coinvolgimento cardiaco, con comparsa in età adulta di cardiomiopatia dilatativa e disturbi di conduzione, associati ad elevato rischio di morte improvvisa (MI) (1).
L’esordio dell’ipostenia avviene solitamente entro l’inizio della seconda decade di vita, a volte preceduto dalla comparsa delle retrazioni tendinee, che possono essere anche gravi ed invalidanti, impattando su postura e cammino (1,2). Al contrario, la forma di distrofia di Emery-Dreifuss legata al cromosoma X (EDMD1), causata da mutazioni nel gene EMD, colpisce solo individui maschi e presenta una distribuzione dell’ipostenia solo omero-peroneale, solitamente preceduta dalle retrazioni tendinee; inoltre, in questa forma l’autonomia del cammino è più a lungo preservata.
La LGMD1B esordisce più tardivamente, solitamente nella terza o quarta decade di vita, e le retrazioni tendinee, in particolari quelle al gomito, sono più tipiche rispetto alla EDMD2 (1,2). Sebbene la nuova classificazione delle Distrofie Muscolari dei Cingoli abbia escluso la forma LGMD1B, tale entità clinica è ancora utile nell’identificare precocemente pazienti mutati in LMNA a causa della specifica distribuzione della debolezza muscolare, prevalente a carico dei muscoli scapolari e pelvici (1,2). Tuttavia, in fase avanzata di malattia, la differenziazione con la EDMD2 può risultare difficoltosa, per il contemporaneo interessamento dei muscoli pelvici in entrambe le forme. La L-CMD è stata riportata in pazienti con mutazioni in LMNA, ed esordio dei sintomi muscolari alla nascita o entro i primi due anni di vita (1,2). Accanto a questa sono state descritte: una grave forma congenita con minimo o assente sviluppo motorio, ed una forma più lieve caratterizzata da prevalente coinvolgimento dei muscoli assiali, normale acquisizione del controllo del capo con successiva insorgenza della“sindrome del capo cadente”, conservata deambulazione (2). Retrazioni tendinee e scoliosi sono molto frequenti. A differenza di EDMD2 e LGMD1B, il coinvolgimento respiratorio è comune, mentre quello cardiaco sembra poco frequente, almeno in fase precoce. Il sistema nervoso centrale non è coinvolto.
Uno studio italiano ha osservato come il fenotipo LGMD1B sia quello più frequente (2), mentre una casistica cinese ha documentato una maggiore frequenza di L-CMD (3).
In generale, la storia naturale delle LMNA-RD è dominata dal coinvolgimento cardiaco e dalle sue complicanze (2,4,5). Generalmente la progressione dell’ipostenia è lenta nel corso degli anni, sebbene impatti sulla qualità di vita e determini disabilità. La perdita della deambulazione avviene comunque in una minoranza dei pazienti (2,4). Un discorso a parte è rappresentato dalle forme più gravi di L-CMD, in cui il coinvolgimento muscolare è molto rilevante, con marcata disabilità motoria (2,3,6).
Coinvolgimento cardiaco
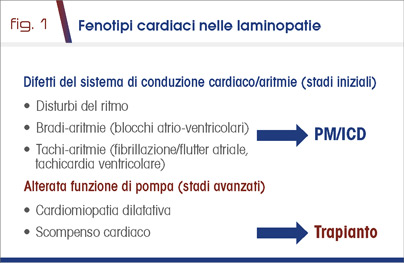 Il coinvolgimento cardiaco è stato riportato nella malattia di Emery-Dreifuss sin dalla sua prima descrizione (7) nel 1966, e confermato nelle LMNA-RD. Esso è caratterizzato da disturbi della conduzione cardiaca sia in senso di bradi-aritmie, per lo più blocchi atrioventricolari (BAV), che di tachi-aritmie - fibrillazione atriale e tachicardia ventricolare - che spesso rappresentano la prima manifestazione della malattia. Il coinvolgimento cardiaco precoce è di solito caratterizzato da un intervallo PR prolungato, che evolve in un BAV avanzato per graduale sostituzione del tessuto di conduzione miocardico con tessuto fibroso e adiposo (5). Quadri di cardiomiopatia dilatativa, con evoluzione verso lo scompenso cardiaco intrattabile e rischio di eventi trombo-embolici, sono propri degli stadi avanzati (Fig. 1). Esistono tuttavia alcune differenze tra le due forme genetiche: nei pazienti con EDMD1 ed in quelli con LGMD1B, di solito il coinvolgimento muscolare scheletrico precede i sintomi cardiaci; nei pazienti EDMD2, l'aritmia cardiaca può essere il primo sintomo che induce a consultare un medico. Anche il tipo e l’età di insorgenza delle aritmie sono diversi: le aritmie atriali sono più frequenti nella EDMD1, quelle ventricolari nelle LMNA-RD (60% vs 3%). Le aritmie ventricolari compaiono in genere più tardivamente nella EDMD1, ma possono insorgere precocemente nelle forme congenite di laminopatia, ed il loro rischio aumenta con l'età. Al contrario i BAV sono di solito più tardivi nelle LMNA-RD (4-6).
Il coinvolgimento cardiaco è stato riportato nella malattia di Emery-Dreifuss sin dalla sua prima descrizione (7) nel 1966, e confermato nelle LMNA-RD. Esso è caratterizzato da disturbi della conduzione cardiaca sia in senso di bradi-aritmie, per lo più blocchi atrioventricolari (BAV), che di tachi-aritmie - fibrillazione atriale e tachicardia ventricolare - che spesso rappresentano la prima manifestazione della malattia. Il coinvolgimento cardiaco precoce è di solito caratterizzato da un intervallo PR prolungato, che evolve in un BAV avanzato per graduale sostituzione del tessuto di conduzione miocardico con tessuto fibroso e adiposo (5). Quadri di cardiomiopatia dilatativa, con evoluzione verso lo scompenso cardiaco intrattabile e rischio di eventi trombo-embolici, sono propri degli stadi avanzati (Fig. 1). Esistono tuttavia alcune differenze tra le due forme genetiche: nei pazienti con EDMD1 ed in quelli con LGMD1B, di solito il coinvolgimento muscolare scheletrico precede i sintomi cardiaci; nei pazienti EDMD2, l'aritmia cardiaca può essere il primo sintomo che induce a consultare un medico. Anche il tipo e l’età di insorgenza delle aritmie sono diversi: le aritmie atriali sono più frequenti nella EDMD1, quelle ventricolari nelle LMNA-RD (60% vs 3%). Le aritmie ventricolari compaiono in genere più tardivamente nella EDMD1, ma possono insorgere precocemente nelle forme congenite di laminopatia, ed il loro rischio aumenta con l'età. Al contrario i BAV sono di solito più tardivi nelle LMNA-RD (4-6).
Diagnosi
La diagnosi di LMNA-RD, sospettata clinicamente sulla base delle caratteristiche menzionate in precedenza, è confermata mediante l’analisi molecolare del gene LMNA (1). Indagini biochimiche quali il dosaggio della creatinchinasi (CK, normale o max 5 volte il limite superiore della norma, più elevato nella L-CMD) o strumentali quali l’elettromiografia, possono aiutare in fase iniziale nella diagnosi differenziale con altre patologie del sistema nervoso periferico, quali patologie del motoneurone e neuropatie; tuttavia, tali esami, se alterati in senso miopatico, non consentono di differenziare le LMNA-RD da altre miopatie.
La risonanza magnetica (RM) dei muscoli di cosce e gambe senza mezzo di contrasto può evidenziare una sostituzione adiposa in muscoli specifici (prevalente nel gastrocnemio mediale e nei vasti, con relativo risparmio del retto femorale), indirizzando l'analisi genetica.
Il quadro istologico muscolare nelle LMNA-RD è generalmente aspecifico, per cui la la biopsia di muscolo non è necessaria nel processo diagnostico dei casi con fenotipo tipico (2).
Diagnosi del coinvolgimento cardiaco
La diagnosi cardiologica si avvale, per la valutazione, di visita cardio-aritmologica, ECG standard e dinamico sec. Holter, ecocardiogramma, ed in casi selezionati, Studio Elettrofisiologico Endocavitario, per la stratificazione del rischio aritmico di MI. La frequenza delle valutazioni (annuale o più frequente) sarà guidata dal giudizio clinico e dalla conoscenza della storia naturale di malattia (4-6,8). La presenza o meno di coinvolgimento muscolare scheletrico, il tipo di aritmie e l’età di insorgenza delle stesse possono essere di aiuto nella diagnosi differenziale tra EDMD1 e EDMD2.
Terapia
Il trattamento dei pazienti si avvale della gestione multidisciplinare di sintomi e complicanze cardiologiche, respiratorie ed ortopediche (1). Sebbene non esiste al momento alcuna terapia causale per le LMNA-RD, terapie promettenti sono in fase di studio preclinico (1).
Terapia del coinvolgimento cardiaco
Si avvale delle stesse indicazioni farmacologiche utilizzate nella popolazione generale, compresa la terapia anticoagulante, in caso di cardiomiopatia dilatativa, fibrillazione atriale ed aritmie (4). Per la prevenzione della morte improvvisa, in caso di bradi-aritmie, di più frequente riscontro nelle EDMD1, è indicato l’impianto di pacemaker (4,9); in caso di tachi-aritmie, di più frequente riscontro nelle laminopatie, trova invece indicazione l’impianto di un defibrillatore (4,10).
La presa in carico del paziente con LMNA-RD presso Centri di riferimento competenti per patologia, necessita di un team multidisciplinare che comprenda anche un aritmologo, in quanto la precoce identificazione dei soggetti ad alto rischio di MI potrebbe salvare loro la vita (9,10).
Bibliografia
- Bonne G, Quijano-Roy S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol 2013, 113, 1367-1376.
- Maggi L, D'Amico A, Pini A, et al. LMNA-associated myopathies: the Italian experience in a large cohort of patients. Neurology 2014, 83, (18), 1634-1644.
- Fan Y, Tan D, Song D, et al. Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients. J Med Genet 2020.
- Wang S., Peng D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019; 60:12–18. doi: 10.1536/ihj.17-60
- Peretto G, Di Resta., Perversi J, et al. Cardiac and Neuromuscular Features of Patients with LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019;171: 458. doi: 10.7326/M18-2768.
- Petillo R, D'Ambrosio P, Torella A, et al. Novel mutations in LMNA A/C gene and associated phenotypes. Acta Myol. 2015 Dec;34(2-3):116-119. PMID: 27199538.
- Emery AE, Dreifuss FE. Unusual type of benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry.1966 Aug;29(4):338-342. doi: 10.1136/jnnp.29.4.338.
- Ben Yaou R, Yun P, Dabaj I, et al. International retrospective natural history study of LMNA-related congenital muscular dystrophy. Brain Commun. 2021 Apr 11;3(3):fcab075. doi: 10.1093/braincomms/fcab075.
- Nigro G, Russo V, Ventriglia VM, et al. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2010 Mar;20(3):174-177. doi: 10.1016/j.nmd.2009.12.004.
- Golzio PG, Chiribiri A, Gaita F. 'Unexpected' sudden death avoided by implantable cardioverter defibrillator in Emery Dreifuss patient. Europace. 2007 Dec;9(12):1158-1160. doi: 10.1093/europace/eum236.