




la Newsletter

Francesca Astra Borruto, Carlo Ferrari, Stefano Mazzoleni, Andrea Zanini, Francesco Macchini
S.C. Chirurgia Pediatrica, GOM Niguarda, Milano
Le malformazioni congenite delle vie aeree polmonari | Includono...
Includono un ampio spettro di alterazioni del parenchima polmonare...
Le malformazioni polmonari congenite (CLM) comprendono alterazioni del parenchima polmonare dovute ad anomalie dello sviluppo broncoalveolare. Le principali forme includono le malformazioni congenite delle vie aeree polmonari (CPAM), che costituiscono dal 30 al 60% delle CLM, il sequestro broncopolmonare (SBP), la cisti broncogena (BC) e l’enfisema lobare congenito (CLO). Esistono forme ibride tra CPAM e SBP. Un'altra malformazione rara è l’atresia bronchiale congenita (1).
Embriogenesi polmonare
Le CLM risultano comprensibili studiando l’organogenesi polmonare, che si sviluppa in cinque fasi. Il tipo e la gravità dell'insulto prenatale influenzano la malformazione. Le CPAM potrebbero derivare da un'alterazione della morfogenesi polmonare o da un’ostruzione focale delle vie respiratorie. L'atresia bronchiale e la BC sono causate da insulti embriogenetici in diverse fasi (2).
Classificazione delle CLM
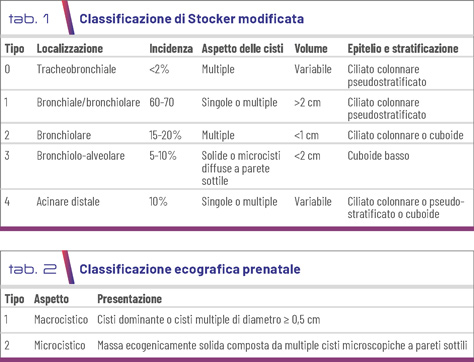 Le CPAM sono masse cistiche che comunicano con l’albero bronchiale, classificate in cinque tipi. Si distinguono in macrocistiche e microcistiche. Nel 1977 Stocker et al. classificarono le CPAM in tre tipi: tipo 1, grandi cisti con cellule mucose; tipo 2, piccole cisti multiple e anomalie associate; tipo 3, forme solide, spesso con shift mediastinico. Successivamente, la classificazione fu aggiornata basandosi sul segmento anatomico dell’albero tracheobronchiale, distinguendo cinque tipi (Tab. 1). Ecograficamente, le CPAM sono classificate in macrocistiche e microcistiche (Tab. 2) (2-3).
Le CPAM sono masse cistiche che comunicano con l’albero bronchiale, classificate in cinque tipi. Si distinguono in macrocistiche e microcistiche. Nel 1977 Stocker et al. classificarono le CPAM in tre tipi: tipo 1, grandi cisti con cellule mucose; tipo 2, piccole cisti multiple e anomalie associate; tipo 3, forme solide, spesso con shift mediastinico. Successivamente, la classificazione fu aggiornata basandosi sul segmento anatomico dell’albero tracheobronchiale, distinguendo cinque tipi (Tab. 1). Ecograficamente, le CPAM sono classificate in macrocistiche e microcistiche (Tab. 2) (2-3).
I SBP sono masse amartomatose di tessuto polmonare non funzionante, non comunicanti con l’albero bronchiale e irrorate da un vaso sistemico aberrante. Le cause sono sconosciute.
Esistono due sottotipi: sequestro intralobare (SI) e sequestro extralobare (SE). I SI, che rappresentano il 75% dei SBP, si sviluppano nella pleura viscerale, principalmente nei lobi inferiori. Hanno prevalentemente un’afferenza arteriosa dalla aorta toracica discendente e drenaggio in atrio sinistro. Sebbene non aerati, presentano una ventilazione collaterale limitata che favorisce complicanze infettive. Il 25% dei SBP è extralobare, con una pleura propria. L’afferenza arteriosa deriva per lo più dall'aorta toraco-addominale e il drenaggio venoso è sistemico, spesso attraverso il sistema azygos o emiazygos o la vena cava verso l'atrio destro. I SE possono causare insufficienza cardiaca congestizia, shunt sinistro-destro o subire torsione del peduncolo vascolare. Le infezioni sono meno comuni poiché non comunicano con l’albero tracheobronchiale. La maggior parte dei SE si trova nella cavità toracica, ma esistono forme sottodiaframmatiche o intradiaframmatiche (3-4).
Le forme miste sono forme ibride con caratteristiche intermedie tra CPAM tipo 1 o 3 e SBP (2).
L’enfisema lobare congenito (CLE) è un'anomalia cartilaginea bronchiale che crea un effetto valvola e provoca sovrainflazione di un lobo polmonare dopo la nascita, rappresentando circa il 10% delle CLM. La sua eziologia è dibattuta e può derivare da fattori intrinseci (assenza di cartilagine, stenosi, broncomalacia) o estrinseci (compressione vascolare). In circa il 50% dei casi è idiopatico. I lobi superiori sinistro e medio destro sono i più colpiti (3).
Le BC sono malformazioni uniloculari della parete posteriore membranosa delle vie aeree. Contengono tessuto cartilagineo, muscolatura liscia e ghiandole bronchiali, e sono rivestite da epitelio colonnare ciliato. Si trovano spesso nel mediastino, sottocarenali, vicine alla trachea o ai bronchi principali, ma possono essere anche intrapolmonari o extratoraciche (collo, addome o cute).
La loro fisiopatologia è sconosciuta. Le cisti mediastiniche, non comunicanti con l’albero tracheobronchiale, contengono fluido o muco e possono causare dispnea comprimendo i bronchi. Le cisti intrapolmonari, collegate all’albero tracheobronchiale, possono causare sintomi respiratori o infezioni per intrappolamento d’aria (3-4).
L'atresia bronchiale congenita è l'interruzione focale di un bronco lobare, segmentale o subsegmentale, spesso associata a mucocele e iperinflazione del segmento polmonare. Il mucocele, causato dall'accumulo di muco per ostruzione, è patognomonico. Il bronco segmentale apicale posteriore del lobo superiore sinistro è il più frequentemente coinvolto. L'atresia bronchiale periferica colpisce bronchi segmentali o subsegmentali e può associarsi a CPAM, SBP e CLE. (3)
Incidenza e malformazioni associate
Negli ultimi anni sembra aumentata l'incidenza delle malformazioni polmonari grazie a migliori diagnostiche prenatali, opzioni terapeutiche fetali e competenze neonatologiche. L'incidenza delle CPAM è circa 1:7.200 nati vivi, ma varia tra i paesi; ad esempio, nel 2015 in Regno Unito era 1:2.500, forse per la mancanza di registri ufficiali. Le CPAM di tipo 1 sono più frequenti (50-70%), seguite dalle CPAM tipo 2 (15-30%) e tipo 3 (5-10%). Le malformazioni associate alle CLM includono malformazioni cardiache (32%) e gastrointestinali (18%). Le cisti broncogene hanno la percentuale più alta di anomalie associate (29%), principalmente vascolari, seguite dalle CPAM (12%), spesso legate a cardiopatie congenite e malformazioni gastrointestinali. SBP è associato a malformazioni gastrointestinali nel 10% dei casi, mentre CLE è collegato a cardiopatie congenite nel 9% dei casi (2-4).
Clinica
La sintomatologia delle CLM è legata alle caratteristiche tissutali di ogni malformazione e all’effetto massa su tessuto polmonare e strutture vicine.
CPAM grandi o CLE possono causare distress respiratorio alla nascita nel 10% dei casi, per shift mediastinico, iperinflazione o pneumotorace (PNX).
Le CPAM, le BC e l’enfisema lobare causano spesso infezioni, che possono essere sintomatiche o asintomatiche, con esiti come fibrosi visibili durante la chirurgia.
Le CLM possono causare atelettasia per effetto massa o atresia bronchiale. Il sintomo principale è la dispnea, associata a desaturazione, stridor, febbre e deterioramento delle condizioni generali.
Il sequestro polmonare può complicarsi con sanguinamento spontaneo o post-trauma, per la vascolarizzazione arteriosa ad alta pressione.
Un'altra complicanza rara del sequestro è lo scompenso cardiaco congestizio.
Nel feto la complicanza più grave è l’idrope, correlata a CPAM e SBP.
Sintomi e complicanze influenzano il trattamento, talvolta richiedendo un intervento precoce. La metà dei pazienti asintomatici sviluppa sintomi nei primi anni, con picco a due anni.
A lungo termine, esiste un rischio neoplastico, con pleuroblastoma nel 2% dei pazienti e possibilità di carcinoma bronchioloalveolare.
La CPAM tipo 4 potrebbe evolvere in pleuroblastoma (PPB) attraverso una mutazione del gene DICER1. Tuttavia, non è dimostrata una relazione specifica tra DICER1 e predisposizione neoplastica nelle CLM. Il 66% dei PPB presenta mutazione germinale DICER1. Mutazioni DICER1 sembrano predisporre ad aumentato rischio di tumori al polmone, rene, ovaio e tiroide (sindrome DICER1). Si stanno studiando altre mutazioni, ad es. a carico del gene KRAS, legate all’espressione MUC5AC, che sono state identificate in cellule mucinose e non mucinose di alcune forme di CPAM (3-5).
Gestione prenatale delle CLM: diagnosi strumentale
Tra la 21° e la 24° settimana, l'ecografia prenatale può identificare malformazioni polmonari come masse ecogene intratoraciche. La sensibilità è del 90% e la specificità del 77%. Nel sequestro polmonare, il Doppler può identificare il vaso anomalo proveniente dal circolo sistemico.
L'ecografia misura il CVR (cystic pulmonary airway malformation volume ratio), che valuta il rischio di idrope fetale, ovvero l’accumulo di liquido nei compartimenti extravascolari e nelle cavità (addome, pericardio, pleura), ad alto rischio di mortalità intrauterina. Un CVR >1.6 indica un rischio elevato di idrope. Il CVR guida anche il follow-up prenatale.
La RM fetale non fornisce informazioni diagnostiche aggiuntive rispetto all'ecografia, ma è utile per casi complessi, ibridi o con diagnosi differenziale di ernia diaframmatica o teratoma mediastinico.
Il picco di crescita delle CPAM è alla 25°-26° settimana. Dalla 28° si può assistere ad arresto della crescita o parziale regressione. In alcuni casi, la malformazione non risulta visibile ai controlli ecografici di fine gestazione.
Le gravidanze con sospette malformazioni devono essere gestite in centri specializzati per diagnosi, parto e l’avvio del percorso terapeutico del nascituro (2-6).
Trattamento fetale
Negli ultimi decenni, il trattamento fetale è diventato cruciale nella gestione delle CLM complicate da idrope, potenzialmente letale. L'ecocardiogramma fetale può indicare segni di outcome sfavorevole come rigurgito valvolare, riduzione della funzione ventricolare o modifiche al Doppler. Solo una piccola parte dei feti con CLM sviluppa complicanze gravi che richiedono interventi prenatali.
Le terapie fetali attuali includono:
- Corticosteroidi sistemici (betametasone), efficaci per CPAM microcistiche con idrope fetale
- Toracentesi
- Shunt toraco-amniotico
- EXIT (EX-utero Intrapartum Therapy): rara, prevede l'intubazione del feto prima dell'interruzione della circolazione placentare e, se necessario, la resezione chirurgica della malformazione
- Ablazione laser del vaso arterioso sistemico aberrante nei SBP, mirata ad interrompere il furto di circolo e ridurre il volume del sequestro (3-4).
Gestione post-natale delle CLM: diagnosi strumentale
La diagnosi dei neonati con sospetto di CLM viene confermata entro il primo mese di vita. Il primo esame è l'RX torace, utile per rilevare PNX e shift mediastinico.
Negli ultimi anni, l’ecografia polmonare ha guadagnato importanza, nonostante sia condizionata dall'esperienza dell'operatore. È economica, priva di radiazioni e utile per valutare versamento pleurico, PNX e lesioni cistiche.
Il gold standard rimane la TC torace con contrasto, che permette una diagnosi accurata e uno studio dettagliato dei rapporti anatomici e vascolari. Nei pazienti sintomatici, la TC e l’intervento sono eseguiti precocemente, mentre negli asintomatici vengono programmati qualche mese dopo la nascita.
Nel primo mese, una RM torace senza contrasto, eventualmente eseguita in sonno spontaneo, può essere utile. È una metodica priva di radiazioni, ma non ancora comparabile alla TC per sensibilità e specificità.
Le immagini devono essere interpretate da radiologi esperti in centri specializzati (1-3,7-9).
Trattamento e indicazioni
La gestione dei neonati con CLM è cambiata notevolmente negli ultimi anni. Le decisioni terapeutiche si basano sulla presenza o assenza di sintomi alla nascita. Il 25% dei neonati presenta distress respiratorio che richiede intervento chirurgico precoce. Ascite, polidramnios e valori CVR > 0.84 sono associati a rischio maggiore di distress respiratorio post-natale. Il 75% dei neonati è asintomatico. La resezione in pazienti asintomatici è controversa. Alcuni optano per un approccio conservativo, dato che le infezioni nelle CLM sono rare (3-10% nei primi cinque anni). Altri preferiscono un intervento precoce per evitare infezioni gravi e complicanze. Circa il 30% dei pazienti con CPAM sviluppa infezioni o PNX, e il 50% delle malformazioni resecate oltre i 6 mesi mostra segni di infezione cronica. Infezioni ripetute rendono la chirurgia più complessa e aumentano morbidità e mortalità. La chirurgia elettiva precoce sfrutta la crescita compensatoria del polmone e riduce il rischio di complicanze infettive. I dati dell’American Pediatric Surgical Association (APSA) suggeriscono di operare entro i 6 mesi per migliorare i risultati respiratori e facilitare la rimozione del drenaggio toracico. Inoltre, non ci sono indicatori chiari per distinguere una CPAM da un blastoma, rendendo l’approccio conservativo rischioso. Solo l’enfisema lobare congenito segmentale asintomatico può essere trattato conservativamente con minor rischio (3-5,10).
Intervento chirurgico
La lobectomia è il trattamento d’elezione per malformazioni polmonari singole (CPAM e ISL). Recentemente, approcci "lung sparing" come segmentectomie e resezioni atipiche sono stati proposti per preservare il parenchima sano. La scelta tra lobectomia e chirurgia "lung sparing" dipende dalla necessità di rimuovere completamente la lesione e preservare il parenchima. Le resezioni atipiche possono comportare complicanze maggiori e una possibile persistenza di malformazione, rendendo la lobectomia una scelta più sicura e definitiva. Tecniche di resezione parziale sono indicate solo per malformazioni multilobari o bilaterali. Per malformazioni extralobari, come sequestri e cisti broncogene, è consigliata l’asportazione della sola malformazione.
Gli interventi sono complessi e ad alto rischio perioperatorio, quindi devono essere eseguiti in centri specializzati con attrezzature adatte e un team esperto. Strumenti come la NIRS (Near-infrared spectroscopy) monitorano l’ossigenazione dei tessuti durante l’operazione. Si posizionano accessi venosi e un cat etere arterioso, e il paziente viene posto in decubito laterale sul lato sano.
La toracotomia “muscle sparing” prevede un’incisione postero-laterale risparmiando i muscoli serrato anteriore e latissimus dorsi, riducendo dolore e rischi di deformità toraciche. Tuttavia, le tecniche mininvasive (MIS) stanno guadagnando popolarità per i loro benefici, come minori complicanze post-operatorie, ridotto dolore e migliori risultati estetici. La MIS, con strumenti sempre più ridotti, è ora possibile anche per pazienti molto piccoli, in centri con esperienza avanzata in chirurgia mininvasiva (3, 11-12).
Gestione post-operatoria
Nei centri specializzati la maggior parte dei pazienti si risveglia dall’anestesia in sala operatoria o sala risveglio; solo alcuni necessitano terapia intensiva. Non sono necessari controlli radiologici precoci per pazienti clinicamente stabili. I pazienti vengono rialimentati precocemente secondo le linee guida ERAS (Enhanced Recovery After Surgery). L'uso del drenaggio toracico e l'analgesia post-operatoria sono ancora dibattuti. Il drenaggio, riservato a casi selezionati, può causare dolore, ridurre la mobilizzazione e aumentare il rischio di infezioni, ma può rilevare complicanze precoci come perdite aeree o sanguinamenti. I miglioramenti nel campo dell'analgesia consentono ormai dimissioni precoci (2°-4° giorno) senza aumentare il rischio di complicanze (11).
Follow-up e outcome
Dopo le dimissioni i pazienti vengono avviati ad un follow-up multidisciplinare in cui chirurghi pediatri, anestesisti, pediatri, fisioterapisti e pneumologi collaborano attivamente per una presa in carico completa, mirata allo studio nel tempo della funzionalità respiratoria residua e all’identificazione di eventuali complicanze precoci o a lungo termine sullo sviluppo della gabbia toracica.
Tra le figure dedicate alla presa in carico delle famiglie e dei bambini con CLM, può essere di grande beneficio anche quella dello psicologo clinico. In diverse realtà cliniche dove è presente tale specialista, il supporto psicologico viene offerto ai genitori dal momento della diagnosi prenatale e lo psicologo accompagna e sostiene il nucleo familiare in tutte le fasi del percorso.
Le tempistiche e le modalità di follow-up clinico, funzionale e radiologico non sono a tutt’oggi chiaramente codificate. Al follow-up a 5-7 anni è stata dimostrata in molti pazienti una capacità polmonare pari a quella dei pazienti non affetti da malformazioni polmonari, particolarmente nei pazienti sottoposti a chirurgia mininvasiva (4).
Conclusioni
Mentre la gestione delle CLM complicate è sostanzialmente standardizzata in età pre- e post-natale, non è ancora da tutti condiviso il percorso delle forme asintomatiche, sia per gli aspetti inerenti alla diagnosi che per il trattamento.
Ad oggi non sono stati effettuati studi randomizzati che supportino mediante evidenze statistiche le preferenze e i risultati dei singoli centri rispetto a timing e tecniche chirurgiche.
Vi è tuttavia un generale consenso degli esperti che anche le CLM asintomatiche, ad eccezione di CLE segmentari, debbano essere resecate prima dell’anno di vita del bambino per la prevenzione di possibili complicanze, anche severe.
È auspicabile che in futuro, con l’incremento della conoscenza della patologia e lo sviluppo di tecniche chirurgiche sempre più sicure ed efficaci, si possa giungere presto a dati definitivi.
Date la rarità e la complessità di questi bambini, le criticità della gestione clinica, l’evoluzione delle tecniche chirurgiche verso approcci sempre meno invasivi ma che richiedono specifiche curve di crescita, la necessità di un approccio multidisciplinare e di un follow-up a lungo termine, è raccomandabile che la gestione dei bambini con CLM avvenga presso centri altamente specializzati.
Bibliografia
- Leblanc C, Baron M, Desselas E, et al. Congenital pulmonary airway malformations: state-of-the-art review for pediatrician’s use. Eur J Pediatr. 2017;176(12):1559-1571.
- Stocker L, Wellesley DG, Stanton MP, et al. The increasing incidence of foetal echogenic congenital lung malformations: an observational study. Prenat Diagn. 2015;35(2):148–153.
- Pederiva F, Rothenberg SS, Hall N, et al. Congenital lung malformations. Nat Rev Dis Primer. 2023;9(1):60.
- Zobel M, Gologorsky R, Lee H, Vu L. Congenital lung lesions. Semin Pediatr Surg. 2019; 28(4):150821.
- Nasr A, Himidan S, Pastor AC, et al. Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg .2010;45(6):1086-9.
- Crombleholme TM, Coleman B, Hedrick H, et al. Cystic adenomatoid mal- formation volume ratio predicts outcome in prenatally diagnosed cystic adenomatoid malformation of the lung. J Pediatr Surg. 2002;37(3):331-8.
- Macchini F. Thoracoscopic resection of congenital pulmonary airway malformations: timing and technical aspects. J Thorac Dis. 2020; 12(8):3944-3948.
- Macchini F, Borzani I, Cavalli S, et al. Thoracoscopic Resection of Congenital Lung Malformation: Looking for the Right Preoperative Assessment. Eur J Pediatr Surg. 2020;30(5):452-458.
- Pio L, Gentilino V, Macchini F, et al. Congenital lung malformations: a nationwide survey on management aspects by the Italian Society of Pediatric Surgery. Pediatr Surg Int. 2024;40(1):53.
- Yamataka A, Koga H, Ochi T, et al. Pulmonary lobectomy techniques in infants and children. Pediatr Surg Int. 2017;33:483-495.
- Bonnard A. Thoracoscopic lobectomy for congenital pulmonary airway malformation: where are we in 2019? Eur J Pediatr Surg. 2020; 30(2):146-149.
- Rothenberg SS, Middlesworth W, Kadennhe-Chiweshe A, et al. Two decades of experience with thoracoscopic lobectomy in infants and children: standardizing techniques for advanced thoracoscopic surgery. J Laparoendosc Adv Surg Tech A. 2015;25(5):423-8.