




la Newsletter

Alessandra Bassotti
Presidio Regionale Lombardo per la sindrome di Ehlers-Danlos, Fondazione IRCCS Ca' Granda-Ospedale Maggiore Policlinico di Milano
Le sindromi di Ehlers-Danlos | Ipermobilità articolare,...
Ipermobilità articolare, iperestensibilità cutanea e fragilità...
Uomo di 23 anni in prima visita presso il Centro di riferimento per la sindrome di Ehlers-Danlos nel maggio 2015. Nato a termine, sviluppo psico-motorio nella norma, pratica attività sportiva regolare (calcio dai 12 ai 17 anni e ballo latino americano a livello agonistico dai 18 ai 22 anni). Riferisce di condurre uno stile di vita normale e di avere riportato una lesione del legamento crociato anteriore all’età di 17 anni durante il gioco del calcio. Fin da piccolo il paziente presenta la tendenza ad ecchimosi sproporzionate e a lento riassorbimento.
A 17 anni viene riscontrata una iniziale varicosità venosa AAII, per cui all’età di 19 anni esegue una safenectomia dx. Durante l’intervento si verifica la lacerazione della cross safeno-femorale, con difficoltà nella sutura, emorragia importante, e conseguente occlusione della vena femorale. All’esame istologico risultano “focolai di sclerosi ed elastosi della parete della vena, dissociante le fibre muscolari, con focali aree di assottigliamento della parete stessa, dove il tessuto muscolare appare quasi totalmente scomparso”. Seguono molteplici visite e consulenze genetiche fino a giungere nel 2014, quando il paziente ha 22 anni di età, alla diagnosi genetica di sindrome di Ehlers-Danlos (EDS) vascolare per presenza in eterozigosi di mutazione c.2284-2A>G del gene COL3A1. All’esame obiettivo presenta cute liscia, vellutata, pastosa, elastica, evidenza di reticolo venoso sottocutaneo agli arti superiori e a livello della regione lombare; importante varicosità nell'arto inferiore dx; cicatrici atrofiche, slargate, a carta di sigaretta e papiracee; caduta della volta plantare in ortostatismo; caviglie non lasse; rotule lievemente lasse in senso latero-mediale, accenno recurvato bilaterale. Il paziente presenta inoltre lassità dei polsi, lassità a livello metacarpofalangeo e interfalangeo, opposizione completa dei pollici; gomiti =180 gradi; spalle: segno del cassetto lievemente positivo bilateralmente; rachide in asse. Non flessione completa del busto. Indice di Beighton 6/9.
 Da allora esegue controlli seriati ogni 12 mesi comprensivi di studio dell’apparato cardiovascolare (Tab. 1) (1).
Da allora esegue controlli seriati ogni 12 mesi comprensivi di studio dell’apparato cardiovascolare (Tab. 1) (1).
Durante gli accertamenti viene riscontrata una lieve insufficienza delle valvole mitralica e tricuspidalica, non significativa a livello emodinamico. Nel 2018, a seguito di una lieve contusione, presenta un ematoma pelvico a cui si associa trombosi venosa profonda della poplitea dx.
Nel corso degli anni il paziente riferisce benessere clinico, pratica palestra 3-4 volte a settimana, solo un peggioramento graduale della dilatazione del lume della vena femorale alla coscia distale, della poplitea e delle vene profonde della gamba (poplitea di 15 mm) che presentano flusso continuo per ipertensione venosa secondaria alla assenza delle vene femorali comuni ed iliache, associato ad incontinenza valvolare. Sempre in corso di accertamenti viene riscontrata splenomegalia.
Il 21 gennaio 2024 il paziente decede improvvisamente mentre sta rientrando a casa: l’autopsia rivela la rottura dell’aorta dal bulbo aortico fino a tutta l’aorta toracica.
Unica terapia consigliata e assunta: celiprololo 100mg aumentato ogni 6 mesi fino al dosaggio di 400mg/die, terapia preventiva (2).
Le sindromi di Ehlers-Danlos
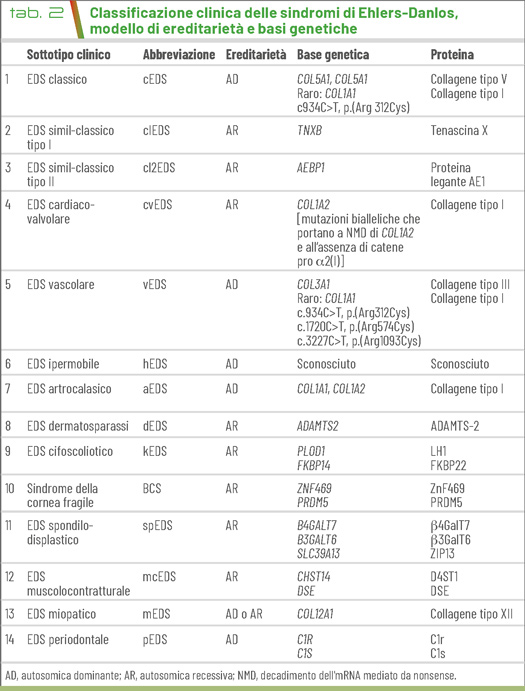 Con il termine "sindrome di Ehlers-Danlos" si intende un gruppo eterogeneo di disordini ereditari del tessuto connettivo caratterizzati da ipermobilità articolare, iperestensibilità cutanea e fragilità generalizzata dei tessuti (3) (Tab. 2). La forma vascolare si trasmette come carattere autosomico dominante, 1/50.000-200.000, data da mutazioni del gene COL3A1, che sintetizza per il collagene tipo III, che si trova a livello della cute, dei vasi sanguigni e degli organi interni. Caratterizzata da aneurismi in giovane età, rotture di organi interni, fistole artero-venose, pneumotoraci spontanei, ecchimosi spontanee, pelle sottile, reticolo venoso, acrogeria, vene varicose, ipermobilità delle piccole articolazioni, facies tipica.
Con il termine "sindrome di Ehlers-Danlos" si intende un gruppo eterogeneo di disordini ereditari del tessuto connettivo caratterizzati da ipermobilità articolare, iperestensibilità cutanea e fragilità generalizzata dei tessuti (3) (Tab. 2). La forma vascolare si trasmette come carattere autosomico dominante, 1/50.000-200.000, data da mutazioni del gene COL3A1, che sintetizza per il collagene tipo III, che si trova a livello della cute, dei vasi sanguigni e degli organi interni. Caratterizzata da aneurismi in giovane età, rotture di organi interni, fistole artero-venose, pneumotoraci spontanei, ecchimosi spontanee, pelle sottile, reticolo venoso, acrogeria, vene varicose, ipermobilità delle piccole articolazioni, facies tipica.
Diagnosi
La diagnosi differenziale si pone con le altre collagenopatie che coinvolgono i vasi, come la sindrome di Marfan, la sindrome di Loeys-Dietz, lo psudoxantoma elasticum, e la sindrome delle arterie tortuose.
La diagnosi è oggi prevalentemente clinica e si basa sulla presenza di criteri diagnostici maggiori e minori, differenziati per ogni tipo di EDS (4-5).
È possibile anche l’indagine genetica attraverso la ricerca di mutazioni nei diversi geni coinvolti, ma solo nella forma vascolare il test genetico riveste un ruolo significativo.
Per evitare eventi vascolari improvvisi e fatali propri dell’EDS vascolare, occorre prendere in considerazione: familiarità positiva, eventi vascolari nel probando, vene varicose precoci e gli altri criteri diagnostici sopra citati come invece non è stato fatto nel caso descritto durante la valutazione delle varici effettuata nella prima struttura.
In quella occasione andava infatti subito indagato il sospetto di forma vascolare per intervenire nel modo corretto ed evitare eventi emorragici, tramite la genetica e gli esami strumentali per lo studio dei vasi, come l’angioTAC torace addome e l’angio-RMN del circolo cerebrale.
Conclusioni
Il riconoscimento precoce di una forma di EDS vascolare può salvare la vita al paziente permettendo di riconoscere prontamente aneurismi o dissecazioni, eventualmente da trattare chirurgicamente, e rappresenta l’opportunità per fornire al paziente importanti indicazioni sullo stile di vita. È infatti necessario che i pazienti, nella vita di tutti i giorni, evitino situazioni potenzialmente pericolose come subire urti e sbalzi pressori o, nel caso pratichino sport ed attività fisiche, scontri o movimenti irregolari come pure accelerazioni/decelerazioni rapide.
Fondamentale, inoltre, che i pazienti vengano monitorati ogni 12 mesi circa: il follow-up permette infatti, per quanto possibile, il riconoscimento di eventuali condizioni cliniche potenzialmente fatali, riducendo ma purtroppo non eliminando il rischio di evento improvviso.
Bibliografia
- Sindrome di Ehlers-Danlos, Percorso Diagnostico Terapeutico e Assistenziale della Rete Regionale per le Malattie Rare - Lombardia, revisione del 2023: https://malattierare.marionegri.it/index.php/pdta-schede
- Baderkhan H, Wanhainen A, Stenborg A, et al. Celiprolol Treatment in Patients with Vascular Ehlers-Danlos Syndrome Eur J Vasc Endovasc Surg. 2021;61(2):326-331.
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26.
- Frank M, Adham S, Seigle S, et al. Vascular Ehlers-Danlos Syndrome: Long-Term Observational Study. J Am Coll Cardiol. 2019;73(15):1948-1957.
- Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Eh-lers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000; 342(10):673-80.