




la Newsletter

Carola Maria Gagliardo, Davide Noto
Dipartimento di Promozione della Salute, Salute Materno-Infantile, Medicina Interna e Specialistica di Eccellenza “G. D’Alessandro” (PROMISE), Università di Palermo
Malattia di Erdheim-Chester: un caso clinico complesso | Il...
Il coinvolgimento multiorgano da parte del danno fibrotico...
Nel luglio 2020 un uomo di 70 anni si presentava al Pronto Soccorso per dolore addominale. L’anamnesi patologica remota era caratterizzata da diabete insipido, ipertensione arteriosa, ipertrofia prostatica benigna, depressione e disturbi della memoria.
La TC dell'addome non mostrava reperti patologici che spiegassero il dolore addominale, mentre al contrario evidenziava lesioni osteosclerotiche della pelvi e della porzione prossimale dei femori, ipertrofia prostatica con densità non uniforme e ispessimento bilaterale della fascia perirenale.
La TC del torace mostrava abbondante versamento pericardico (spessore 5 cm), trattato con pericardiocentesi in urgenza. La TC con contrasto e la RMN dell'addome e della pelvi confermavano i reperti descritti.
La scintigrafia ossea marcata con tecnezio-99m evidenziava un'intensa captazione bilaterale della diafisi omerale destra, della testa omerale sinistra, delle diafisi femorali e tibiali bilateralmente e delle creste iliache. La 18-FDG PET confermava le stesse aree ossee di iperaccumulo. Gli esami ematochimici mostravano lieve anemia ipocromica-normocitica, lieve incremento di VES e PCR, PSA libero e totale nella norma. I mesi successivi il paziente iniziava a lamentare un dolore osseo diffuso.
Le radiografie di omeri, femori, gambe e bacino documentavano aree di iperdiafania circondate da osteosclerosi. Il periostio appariva ispessito. Tali lesioni hanno fatto sorgere il sospetto di carcinoma prostatico metastatizzante le ossa o di malattia di Paget. Tre biopsie ossee hanno tuttavia mostrato reperti non diagnostici.
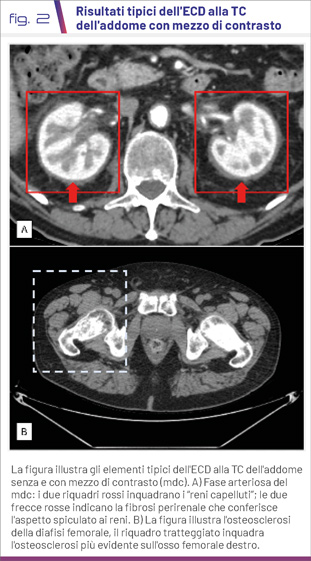
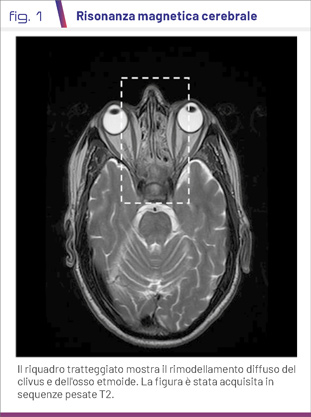 Il paziente è giunto nel nostro ambulatorio di Malattie Metaboliche Rare dell’AOUP Policlinico “Paolo Giaccone” nel marzo 2021 per un sospetto di osteopatia metabolica. Alla visita sono stati notati xantelasmi palpebrali. La RMN cerebrale ha mostrato un diffuso rimodellamento osteostrutturale del clivus, dell'osso etmoide e dei seni mascellari, ma non ha documentato lesioni parenchimali nell'ipofisi compatibili con il diabete insipido (Fig. 1). Una nuova TC torace-addome con mezzo di contrasto ha mostrato tessuto fibroso perirenale, lungo la fascia para-renale anteriore e posteriore, con aspetto spiculato, come da “reni capelluti”, esteso al tratto ureterale prossimale con un parziale effetto compressivo (Fig. 2A). Sono state descritte anche un'osteosclerosi delle ossa lunghe del bacino e dei segmenti metadiafisari di femori e omeri bilateralmente (Fig. 2B). È stata quindi sospettata diagnosi clinica di malattia di Erdheim-Chester (ECD). La biopsia cutanea degli xantelasmi di maggio 2021 ha documentato l'infiltrazione di istiociti schiumosi CD68 (+), Cd1a (-), BRAFV600E (-) (1). Nonostante i risultati negativi dell'analisi immunoistochimica, è stata eseguita valutazione genetica presso l’Istituto Meyer di Firenze, che ha identificato la mutazione BRAFV600E.
Il paziente è giunto nel nostro ambulatorio di Malattie Metaboliche Rare dell’AOUP Policlinico “Paolo Giaccone” nel marzo 2021 per un sospetto di osteopatia metabolica. Alla visita sono stati notati xantelasmi palpebrali. La RMN cerebrale ha mostrato un diffuso rimodellamento osteostrutturale del clivus, dell'osso etmoide e dei seni mascellari, ma non ha documentato lesioni parenchimali nell'ipofisi compatibili con il diabete insipido (Fig. 1). Una nuova TC torace-addome con mezzo di contrasto ha mostrato tessuto fibroso perirenale, lungo la fascia para-renale anteriore e posteriore, con aspetto spiculato, come da “reni capelluti”, esteso al tratto ureterale prossimale con un parziale effetto compressivo (Fig. 2A). Sono state descritte anche un'osteosclerosi delle ossa lunghe del bacino e dei segmenti metadiafisari di femori e omeri bilateralmente (Fig. 2B). È stata quindi sospettata diagnosi clinica di malattia di Erdheim-Chester (ECD). La biopsia cutanea degli xantelasmi di maggio 2021 ha documentato l'infiltrazione di istiociti schiumosi CD68 (+), Cd1a (-), BRAFV600E (-) (1). Nonostante i risultati negativi dell'analisi immunoistochimica, è stata eseguita valutazione genetica presso l’Istituto Meyer di Firenze, che ha identificato la mutazione BRAFV600E.
 È stata quindi praticata la terapia mirata con vemurafenib (BRAF-inibitore), previa approvazione del comitato etico dell'ospedale.
È stata quindi praticata la terapia mirata con vemurafenib (BRAF-inibitore), previa approvazione del comitato etico dell'ospedale.
Due settimane dopo il farmaco è stato sospeso a causa della comparsa di un rash eritemato-papulare diffuso sul tronco e sugli arti, come da scheda tecnica del farmaco (2). L'eruzione è regredita in pochi giorni con una terapia a base di steroidi e antistaminici, ma il paziente ha rifiutato di riprendere il farmaco.
ECD: descrizione clinica
Il caso presentato mostra molte delle manifestazioni cliniche della ECD, come lesioni ossee osteosclerotiche, diabete insipido, versamento pericardico e fibrosi perirenale. L'osteosclerosi bilaterale delle ossa lunghe alla metadiafisi rappresenta la manifestazione più frequente della ECD, interessando l'80-95% dei pazienti (3). Le lesioni ossee possono essere rilevate mediante raggi X, TC, RMN, scintigrafia ossea o PET. La scintigrafia ossea con 99mTc-MDP è utile per differenziare la ECD da altre malattie sclerotiche. Le diagnosi alternative da prendere in considerazione includono osteomielite, malattia di Paget, malattia di Grave, linfoma, sarcoidosi, metastasi e malattie da accumulo di lipidi.
L'infiltrazione istiocitaria dello spazio retroperitoneale nell'ECD porta spesso a un ispessimento bilaterale diffuso dei tessuti molli perirenali con un pattern definito “rene capelluto o hairy kidney”, che è altamente suggestivo di malattia (4). Il 44% dei pazienti con coinvolgimento peri-renale sviluppa nel tempo insufficienza renale.
Tra i disturbi del sistema nervoso centrale si annoverano le sindromi cerebellari e piramidali, crisi epilettiche, cefalee, deficit cognitivi, paralisi dei nervi cranici, segni neuropsichiatrici. Il diabete insipido centrale rappresenta la prima manifestazione clinica della ECD nel 25-48% dei casi e può svilupparsi diversi anni prima della diagnosi (5).
La malattia del pericardio è un reperto tipico dell'ECD. Il versamento massivo, come nel nostro caso, è raro (6). Manifestazioni cutanee, quali xantelasma palpebrale e lesioni papulo-nodulari, malattie interstiziali polmonari, della tiroide, mammella e linfonodi possono pure presentarsi (7).
Diagnosi
La diagnosi richiede sia elementi clinici sia reperti istopatologici (8). I segni istopatologici della ECD consistono nell'infiltrazione dell'organo da parte di istiociti schiumosi o carichi di lipidi, insieme a fibrosi e cellule di Touton. L'immunoistochimica mostra un profilo caratteristico delle cellule ECD: CD68, CD163 e fattore XIIIa positivi, CD1a e CD207 negativi, S-100 (+) (9). L'analisi immunoistochimica per la colorazione BRAFV600E rileva la proteina mutante nel 50% dei casi. Tuttavia, nei casi di immunoistochimica negativa è preferibile eseguire indagini genetiche per confermare/rilevare la mutazione corretta, come è accaduto nel nostro paziente (5).
Trattamento
Le terapie attuali sono personalizzate in base al rilevamento di mutazioni specifiche. La Food and Drug Administration (FDA) ha approvato vemurafenib come terapia di prima linea per i pazienti portatori della mutazione BRAFV600E, mentre l'Agenzia Europea dei Medicinali (EMA) non ha approvato il trattamento, che viene quindi somministrato off-label (10).
Tra gli eventi avversi di vemurafenib possono presentarsi cancro della pelle (carcinoma a cellule squamose, melanoma o altri), reazioni di ipersensibilità cutanea, disturbi epatici e renali, sindrome di Stevens-Johnson e necrolisi epidermica tossica (11, 12). Il nostro paziente ha mostrato un rash cutaneo maculo-papulare dopo due settimane dall'inizio della terapia, regredito con gli steroidi. Secondo la scheda tecnica, in questi casi il farmaco deve essere temporaneamente sospeso (2). Nel nostro caso, il farmaco è stato sospeso definitivamente per scelta personale del paziente.
Cobimetinib, inibitore di MEK, rappresenta un'alternativa terapeutica a vemurafenib, poiché più del 25% dei pazienti presenta anche mutazioni attivanti la via MAPK (MAP2K1, KRAS e NRAS) diverse da BRAFV600E.
Questo farmaco ha dimostrato di essere sicuro ed efficace nella stabilizzazione della malattia in pazienti con mutazione BRAFV600E che hanno manifestato eventi avversi (tra cui la sindrome DRESS) dopo il trattamento con vemurafenib (13). Il nostro paziente ha comunque rifiutato questa opzione terapeutica.
Conclusioni
Il presente caso clinico evidenzia come la diagnosi di ECD possa essere sfuggente. Il contemporaneo coinvolgimento multiorgano da parte del danno fibrotico associato al rimodellamento osseo dovrebbe presto far sospettare l'esistenza di un'unica patologia a cui ricondurre tutte le manifestazioni. Inoltre, la contemporanea presenza di versamento pericardico, diabete insipido, fibrosi retroperitoneale e dolore dovrebbe far sorgere il sospetto di ECD.
La divulgazione e quindi la conoscenza di queste rare entità cliniche può permettere al clinico di fare la diagnosi corretta. Una diagnosi precoce, seguita da una terapia tempestiva, è infatti essenziale per modificare la storia naturale della malattia.
Bibliografia
- Costa IBSDS, Costa FAS, Bittar C, et al. Cardiac Tamponade as the First Manifestation of Erdheim-Chester Disease. JACC CardioOncol. 2020;2(2):324-328.
- https://www.ema.europa.eu/en/documents/product-information/zelboraf-epar-product-information_it.pdf
- Goyal G, Young JR, Koster MJ, et al. The Mayo Clinic Histiocytosis Working Group. The Mayo Clinic Histiocytosis Working Group Consensus Statement for the Diagnosis and Evaluation of Adult Patients With Histiocytic Neoplasms: Erdheim-Chester Disease, Langerhans Cell Histiocytosis, and Rosai-Dorfman Disease. Mayo Clin Proc. 2019; 94(10):2054-2071.
- Dion E, Graef C, Haroche J, et al. Imaging of thoracoabdominal involvement in Erdheim-Chester disease. 2 AJR Am J Roentgenol. 2004;183(5):1253-60.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016; 127(22):2672-81.
- Yoon M, Lee SH, Shim HS, Kang SM. Erdheim-Chester disease presenting as an intracardiac mass and pericardial effusion confirmed by biopsy: a case report. Eur Heart J Case Rep. 2021;5(10):ytab351.
- Papo M, Emile JF, Maciel TT, et al. Erdheim-Chester Disease: a Concise Review. Curr Rheumatol Rep. 2019;21(12):66.
- Haroche J, Cohen-Aubart F, Amoura Z (2020: Erdheim-Chester disease. Blood. 2020;135(16):1311-1318.
- Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124(4):483-92.
- Cohen Aubart F, Emile JF, Carrat F, et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood. 2017;130(11):1377-1380.
- Brychtová M, Vlachová M, Gregorová J, et al. Erdheim-Chester disease. Klin Onkol. 2021; 34(6):434-439.
- Starkebaum G, Hendrie P. Erdheim-Chester disease. Best Pract Res Clin Rheumatol. 2020;34(4):101510.
- Cohen Aubart F, Emile JF, Maksud P, et al. Efficacy of the MEK inhibitor cobimetinib for wild-type BRAF Erdheim-Chester disease. Br J Haematol. 2018; 180(1):150-153.