




la Newsletter

Alessandro Innocenti, Francesca Lanzani, Elio Clemente Agostoni
SC Neurologia e Stroke Unit, ASST Grande Ospedale Metropolitano Niguarda, Milano
Miastenia gravis | La miastenia gravis è una rara patologia...
La miastenia gravis è una rara patologia autoimmune...
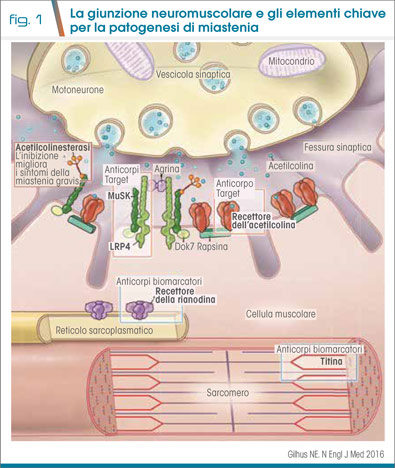 La miastenia gravis (MG) è una patologia autoimmune caratterizzata da una alterata trasmissione del segnale nervoso nella giunzione neuromuscolare (NMJ). Essa è causata da autoanticorpi diretti contro i recettori post-sinaptici della acetilcolina (AChR) o contro proteine funzionalmente correlate ad essi; tali anticorpi (Ab) alterano la normale trasmissione neuromuscolare (NMT), determinando debolezza muscolare e affaticabilità che prevalgono in specifici distretti muscolari e hanno un andamento caratteristicamente fluttuante (Fig.1).
La miastenia gravis (MG) è una patologia autoimmune caratterizzata da una alterata trasmissione del segnale nervoso nella giunzione neuromuscolare (NMJ). Essa è causata da autoanticorpi diretti contro i recettori post-sinaptici della acetilcolina (AChR) o contro proteine funzionalmente correlate ad essi; tali anticorpi (Ab) alterano la normale trasmissione neuromuscolare (NMT), determinando debolezza muscolare e affaticabilità che prevalgono in specifici distretti muscolari e hanno un andamento caratteristicamente fluttuante (Fig.1).
Epidemiologia e patogenesi
La malattia può presentarsi a qualsiasi età. Studi epidemiologici (1) hanno riportato una incidenza di 8-10 casi su milione, influenzata da sesso ed età: in età giovanile, intorno ai 30 anni, le donne sono colpite 3 volte di più rispetto agli uomini; intorno ai 50 anni, periodo in cui vi è un secondo picco di incidenza, gli uomini sono maggiormente colpiti.
In circa il 15% dei pazienti la MG si associa ad altre patologie autoimmuni (es. distiroidismi, lupus eritematoso sistemico, artrite reumatoide), soprattutto nei giovani con iperplasia timica (vedi oltre).
Se è noto il meccanismo con cui gli Ab anti-AChR determinano un danno complemento-mediato sulla membrana postsinaptica, con endocitosi recettoriale e sostanziale blocco funzionale della NMT, meno chiaro è il meccanismo alla base della perdita di tolleranza. È probabile che esso sia multifattoriale, legato a fattori genetici, epigenetici, ambientali ed alla modulazione tra immunità adattativa ed innata (2). Il timo è un organo checkpoint chiave nel determinare l’attivazione della risposta autoimmune AChR-Ab mediata: nel timo sono presenti subunità di AChR che fungono da antigeni, e in particolare nei pazienti con iperplasia timica sono state documentate l'attivazione aberrante del sistema immunitario e la specifica sensibilizzazione verso AChR.
È inoltre probabile che fattori ambientali (ad es. infezioni da Epstein Barr o Parvovirus B19) agiscano da trigger per la risposta autoimmune.
Probabilmente diversa è la patogenesi della MG da Ab diretti contro altre proteine della membrana, quali la tirosinchinasi muscolo-specifica (MuSK) e la proteina LRP4 (Low-density lipoprotein receptor-related protein 4): esse sono funzionalmente legate ad AChR ed una loro disfunzione comporta una ridotta concentrazione dei recettori sulla membrana postsinaptica.
Clinica e varianti
I tratti clinici caratteristici della MG sono debolezza muscolare ed affaticabilità che tipicamente peggiorano con l’esercizio e fluttuano in intensità, frequentemente con peggioramento serotino. Il pattern dei muscoli interessati è spesso distintivo: la muscolatura oculare è frequentemente coinvolta e ptosi o diplopia sono il sintomo iniziale nei 2/3 dei casi.
Entro i primi anni dall’esordio la maggioranza dei pazienti sviluppa un coinvolgimento muscolare generalizzato, con interessamento della muscolatura artuale (generalmente prossimale, con difficoltà nel sollevare le braccia o fare le scale) o assile (es. flessori del collo).
Nella forma generalizzata si può avere un coinvolgimento anche della muscolatura bulbare, che nel 10-15% dei pazienti può essere il sintomo di esordio: rinolalia, ipofonia, disfagia e difficoltà masticatorie sono secondarie ad affaticabilità precoce della muscolatura orofaringea e masticatoria; alcuni pazienti possono sviluppare dispnea per coinvolgimento della muscolatura respiratoria. In una minoranza di pazienti la dispnea può rapidamente progredire verso una insufficienza respiratoria acuta (crisi miastenica), che può essere scatenata da fattori esterni quali infezioni, farmaci o interventi chirurgici. La crisi miastenica è una urgenza neurologica che può determinare la necessità di supporto ventilatorio e terapie quali immunoglobuline o plasmaferesi (vedi oltre).
Il decorso della malattia è variabile, ma generalmente progressivo. La malattia ha fasi di spontanea remissione e fasi di recrudescenza. L’andamento, comunque, è secondario ad una serie di elementi quali gli autoAb presenti, l’assetto del timo, il fenotipo di malattia, le patologie concomitanti e le terapie. L’analisi combinata di queste caratteristiche cliniche e non cliniche permette di identificare delle varianti e di definire la prognosi (3) (Tab.1).

Sebbene molti pazienti inizino con sintomi oculari, solo in circa il 15% dei casi la MG resta unicamente oculare. Nei pazienti con miastenia oculare gli autoAb (anti-AChR o più raramente anti-LRP4, mai anti-MuSK) sono presenti in circa il 50% dei casi e correlano con un aumentato rischio di generalizzazione.
Nei pazienti con MG generalizzata e Ab anti-AChR si identificano 2 gruppi in base all’età di esordio. Nei pazienti early onset la malattia esordisce prima dei 50 anni; la forma è più frequente nelle donne e in associazione a iperplasia timica. Nei pazienti late onset la malattia esordisce invece dopo i 50 anni: i pazienti sono in genere maschi, con atrofia timica. In tali pazienti possono essere presenti anche altri autoAb che non hanno un ruolo patogenico nella malattia ma fungono da biomarcatori: gli Ab antititina e antirianodina sono diretti contro proteine intracellulari nella NMJ, si associano ad un fenotipo severo e spesso alla presenza di timoma (presente nel 10-15% dei casi di MG); altri autoAb (es. Kv1.4) non sembrano avere un ruolo prognostico.
A conferma di un diverso meccanismo eziologico, le forme di MG con autoAb anti-MuSK e anti-LRP4 non si associano a malattia timica. Le forme anti-MuSK (1-10% dei casi) sono prevalenti nel sesso femminile e si associano a forme severe, con precoce coinvolgimento bulbare e necessità di terapia immunosoppressiva. Nelle forme anti-LRP4 (1-3%) i sintomi sono invece lievi.
Vi sono infine pazienti affetti da MG nei quali non vengono trovati Ab (sieronegativi), probabilmente per un titolo anticorpale sotto la sensibilità diagnostica o per Ab non ancora identificati.
Diagnosi
La fluttuazione dei sintomi, l’affaticabilità, il peculiare pattern muscolare spesso consentono di porre diagnosi di MG già su base clinica.
La positività degli autoAb anti-AChR, anti-MuSK o anti-LRP4 consentono di confermare la diagnosi, sebbene una loro negatività non permetta di escluderla.
Lo studio elettromiografico è particolarmente importante quando la clinica o la sierologia non siano dirimenti. La stimolazione ripetitiva è il test più utilizzato, con caratteristica risposta decrementale progressiva nelle ampiezze dei potenziali di azione muscolare registrati. L’elettromiografia a singola fibra è il test più sensibile e nei pazienti affetti mostra un aumento dell’intervallo temporale nell’attivazione di due fibre adiacenti (jitter) in alcuni distretti muscolari.
Infine è importante determinare l’assetto timico con imaging mediastinico per valutare l’eventuale presenza di timoma o di iperplasia timica.
Terapia
L’obiettivo della terapia della MG è la remissione della sintomatologia o l’assenza di sintomi funzionalmente invalidanti (4).
A tale scopo il trattamento integra diversi approcci. Le terapie comprendono principalmente trattamenti sintomatici (inibitori dell’acetilcolinesterasi), la timectomia e l’immunoterapia sia con immunosoppressori che, più recentemente, con farmaci biologici.
Tra i sintomatici, il farmaco più utilizzato è la piridostigmina. La risposta varia a seconda del sottogruppo; il dosaggio va modulato in base alla risposta clinica e agli effetti collaterali (es. gastrointestinali). Spesso tuttavia tale terapia non consente un controllo dei sintomi e sono necessarie terapie immunosoppressive.
Lo steroide (es. prednisone) rimane nella maggior parte dei casi la prima linea. L’uso deve essere tuttavia cauto, sia per il rischio di un iniziale e transitorio peggioramento sia soprattutto per gli effetti collaterali.
Per minimizzare l’uso di steroide sono essenziali gli immunosoppressori (5); in assenza di ampi studi comparativi, la scelta del farmaco è spesso basata sull’esperienza. Azatioprina e micofenolato mofetile sono le terapie più utilizzate, con effetti collaterali in genere ben tollerati ma con lunga tempistica di azione (circa 6 mesi con azatioprina).
Qualora, come nella crisi miastenica, sia necessaria una terapia urgente, le migliori opzioni sono la plasmaferesi o le immunoglobuline ev (6).
Il 10% circa dei pazienti rimane comunque refrattario ai trattamenti; tale scenario è tuttavia cambiato negli ultimi anni con i farmaci biologici (7) (Fig. 2). Tra questi, eculizumab (8) (primo farmaco approvato da FDA ed EMA per la MG refrattaria) è un anticorpo monoclonale diretto contro il complemento C5. Rituximab, un anticorpo anti-CD20, è efficace in particolare nelle forme anti-MuSK. Efgartigimod, recentemente approvato sia da FDA che da EMA, agisce come antagonista del recettore Fc neonatale e riduce gli autoAb circolanti.
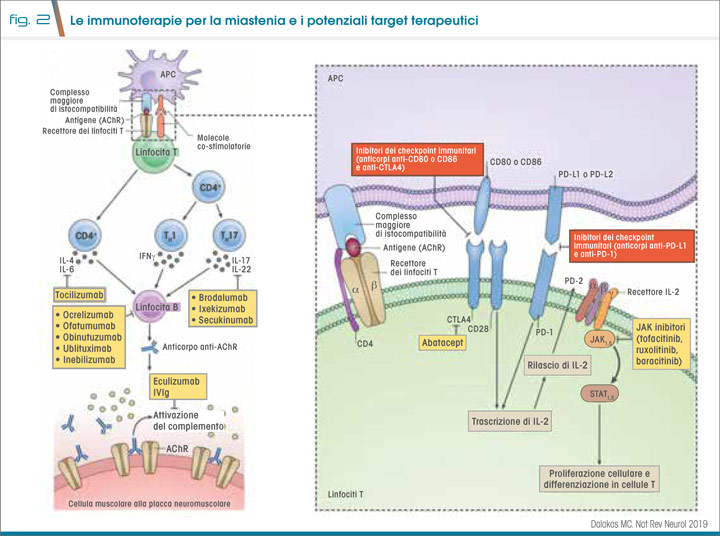
Nuove direzioni
L’uso dei biologici, di cui molti in fase di sviluppo, permette concrete possibilità di trattamento personalizzato, in particolare nei pazienti con MG refrattaria. I costi importanti di tali terapie richiedono comunque riflessioni attente sulle indicazioni.
Bibliografia
- Carr AS, Cardwell CR, McCarron PO et al. A systematic review of population based epidemiological studies in Myasthenia Gravis. BMC Neurol. 2010;10:46.
- Mantegazza R, Bernasconi P, Cavalcante P. Myasthenia gravis: from autoantibodies to therapy. Curr Opin Neurol. 2018;31(5):517–25.
- Gilhus NE. Myasthenia Gravis. N Engl J Med. 2016;375(26):2570–81.
- Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021;96(3):114–22.
- Lascano AM, Lalive PH. Update in immunosuppressive therapy of myasthenia gravis. Autoimmun Rev. 2021;20(1):102712.
- Gajdos P, Chevret S, Toyka KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev 2012;12:CD002277.
- Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol 2019;15(2):113–24.
- Howard JF, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 2017;16(12):976–86.