




la Newsletter

Marianna Alagia, Simona Fecarotta
Dipartimento Materno-Infantile, Azienda Ospedaliera Universitaria Federico II, Napoli
Nuove frontiere diagnostiche nella malattia di Pompe: lo screening...
La terapia enzimatica sostitutiva ha modificato in modo...
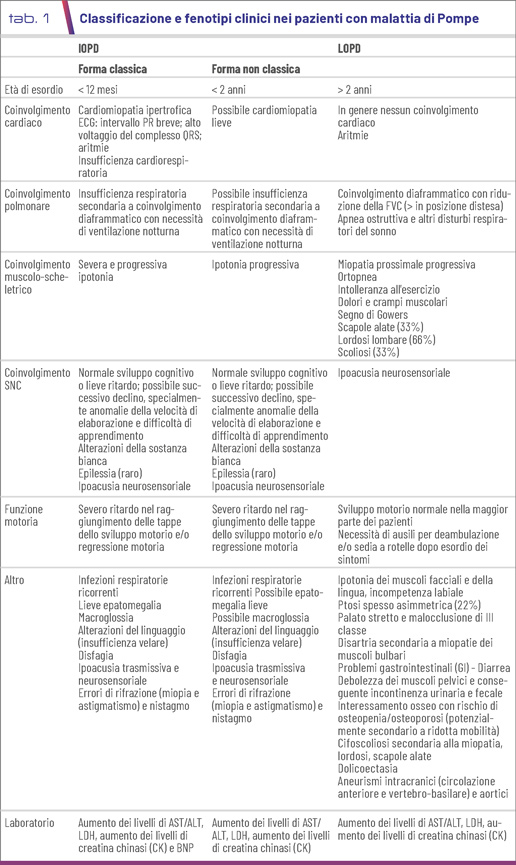
La malattia di Pompe (MP) è una malattia autosomica recessiva con un’incidenza stimata di 1:40.000 - 1:146.000, causata da un difetto dell'a-glucosidasi acida lisosomiale (GAA), che determina l'accumulo generalizzato di glicogeno lisosomiale, con una sintomatologia progressiva a carico dei muscoli scheletrici e cardiaco (1). La malattia è caratterizzata da un ampio spettro fenotipico; si distinguono classicamente forme a esordio infantile (IOPD), le più gravi, che nei primi mesi di vita presentano cardiomiopatia ipertrofica, ipotonia muscolare progressiva ed una prognosi infausta con morte precoce per insufficienza cardiorespiratoria entro i primi 1-2 anni di vita, e forme a esordio tardivo (LOPD), che si possono manifestare a qualsiasi età con una debolezza muscolare progressiva (Tab. 1). L’impatto clinico e l’età di esordio sono determinati principalmente dall’attività enzimatica GAA residua. I pazienti IOPD presentano un’attività enzimatica residua molto ridotta (<3%) o assente mentre nei pazienti LOPD l’attività della GAA è variabilmente ridotta (3-30%), con una corrispondente ampia variabilità delle manifestazioni cliniche, dell’età di insorgenza e decorso (2).
La diagnosi viene stabilita dalla evidenza di un deficit dell’attività GAA confermata dall'analisi molecolare del gene. Il tetrasaccaride urinario (Glc4), derivato dalla degradazione del glicogeno, è un biomarcatore specifico di accumulo, nel contesto della ridotta attività GAA.
La terapia enzimatica sostitutiva (ERT) con GAA umano ricombinante (rhGAA) ha modificato in modo significativo la storia naturale della malattia. La risposta favorevole alla terapia è fortemente dipendente dall’inizio precoce della stessa. Nella IOPD, la ERT deve essere iniziata il prima possibile poiché ritardi anche di giorni possono influenzare i risultati. Nei pazienti con LOPD la ERT è associata a risultati migliori se iniziata prima che si verifichi un danno muscolare irreversibile (3,4).
La rarità della MP e la sovrapposizione delle manifestazioni cliniche con altre condizioni determinano un importante ritardo diagnostico, per cui lo stato funzionale del paziente è spesso già gravemente compromesso al momento della diagnosi. Lo screening neonatale (SN) sembra essere la migliore strategia per l’identificazione precoce e presintomatica dei pazienti affetti, soprattutto nei casi IOPD, ed in assenza di una storia familiare.
Screening neonatale per la malattia di Pompe
I principali fattori che supportano l’attuazione di programmi di SN per la MP includono lo sviluppo di nuove promettenti opzioni terapeutiche, i progressi nelle tecnologie, oltre che la spinta emergente da parte di portatori di interesse; tuttavia l’istituzione diffusa del programma rimane impegnativa e per alcuni aspetti controversa. I programmi di SN per la MP sono già attivi in diversi paesi (ad esempio, negli Stati Uniti, Taiwan, Giappone) (5-7) e la malattia di Pompe è stata aggiunta al pannello di screening universale raccomandato (Recomended Uniform Screening Panel, RUSP) dagli Stati Uniti nel 2015 (7). In Italia la malattia di Pompe è oggetto di screening neonatale soltanto in alcune regioni, ma è possibile che tale programma venga esteso nel prossimo futuro sull'intero territorio nazionale.
La proposta di attuazione di programmi di SN per la MP è basata sulla possibilità tecnica di misurare l'attività della GAA su gocce di sangue essiccato su carta bibula (dried-blood-spot, DBS), con diverse metodiche (spettrometria di massa tandem, fluorimetria, microfluidica digitale) (8) e sulla disponibilità dal 2006 di una ERT, e di ulteriori enzimi di nuova generazione recentemente approvati al commercio o prossimi alla approvazione.
Dal punto di vista strettamente tecnico esistono alcune difficoltà che vanno considerate nella implementazione dei programmi, quali la degradazione dell’enzima esposto a temperature non controllate durante il trasporto dei DBS dal punto nascita al laboratorio centralizzato per lo screening, la possibile diluizione del campione esposto ad elevata umidità, l’incapacità del test di distinguere le pseudodeficienze, l’assenza di biomarcatori affidabili, misurabili su DBS. Per quanto considerato, l’analisi contemporanea di più enzimi lisosomiali può essere di aiuto nella determinazione della qualità del campione, evidenziando la riduzione aspecifica di più attività enzimatiche in campioni leucodepleti o esposti a calore e umidità elevati (8), mentre l’analisi genetica rimane fondamentale per il riconoscimento delle pseudodeficienze (varianti alleliche che riducono l'attività della GAA ma che non causano la MP).
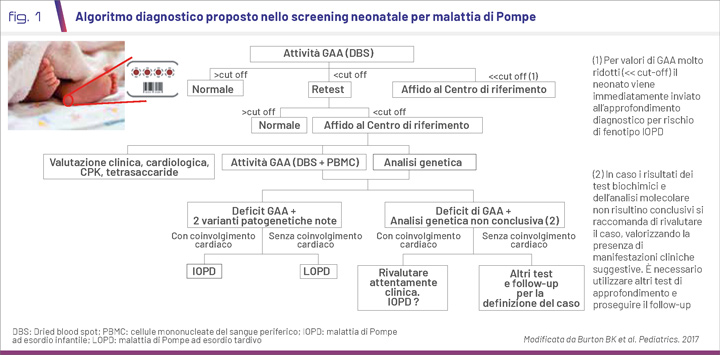
È opportuno ricordare che il dosaggio enzimatico su DBS rappresenta solo un test di screening e non è sufficiente per una diagnosi definitiva (9) (Fig. 1).
Presa in carico, conferma diagnostica, management e follow-up
A seguito della positività allo screening neonatale, il neonato deve essere affidato al Centro clinico di riferimento dello screening per la presa in carico, la conferma diagnostica e la valutazione del fenotipo, attraverso l’integrazione di differenti elementi clinici, biochimici e molecolari (Fig. 1).
La diagnosi deve essere confermata dal dosaggio enzimatico della GAA in almeno uno dei seguenti omogenati cellulari: linfociti del sangue periferico, colture di fibroblasti da biopsia cutanea, biopsia muscolare. La possibilità di una pseudodeficienza di GAA deve essere presa in considerazione per l'interpretazione del test biochimico (10) e l’utilizzo di specifici substrati e tessuti per l’analisi può permettere una migliore risoluzione tra pazienti affetti e non affetti (11).
L'analisi molecolare del gene GAA dovrebbe seguire o accompagnare il saggio enzimatico per la conferma diagnostica e il successivo counseling genetico-familiare. È stata descritta una correlazione genotipo-fenotipo che può, inoltre, fornire informazioni utili per la valutazione del fenotipo e della prognosi (1).
La combinazione dell’analisi dell’attività enzimatica e della conferma genetica rappresenta il gold standard diagnostico. In rari casi non conclusi dai metodi di sequenziamento convenzionale o di nuova generazione possono essere necessari metodi molecolari aggiuntivi per identificare le varianti, come test di splicing genetico, MLPA, analisi di minigeni, analisi di array SNP e sequenziamento Sanger mirato (12).
Per la valutazione del fenotipo ed il riconoscimento dei pazienti IOPD appare importante eseguire una valutazione clinica completa, cardiologica con ECG ed ecocardiogramma, oltre che il dosaggio di alcuni biomarcatori riconosciuti di danno muscolare (CK, AST, ALT, LDH nel sangue) e di accumulo quale il tetrasaccaride (Glc4) nelle urine.
Nei pazienti con fenotipo IOPD i risultati dell’analisi molecolare e la valutazione dello stato CRIM (Cross-Reactive Immunologic Material) dovranno essere disponibili in tempi molto rapidi, tali da consentire la tempestiva introduzione della ERT in associazione ad un opportuno protocollo di immunomodulazione, mentre il riconoscimento presintomatico di pazienti LOPD in età neonatale garantisce la definizione di un follow-up periodico mirato e l’inizio della terapia all’insorgenza dei primi sintomi.
I benefici dei programmi di screening neonatale
Il vantaggio principale dello SN per la MP è la possibilità di ridurre l’odissea diagnostica, soprattutto nei pazienti IOPD, ed introdurre la ERT prima di danni tissutali irreversibili.
Lo SN fornisce inoltre vantaggi secondari relativi alla conoscenza del rischio riproduttivo in coppie di portatori, consentendo scelte informate in merito alla futura pianificazione familiare; alla migliore comprensione della reale prevalenza di malattia, che appare meno rara di quanto finora riportato ed alla maggiore comprensione della storia naturale nelle forme LOPD.
Questioni aperte
L’identificazione dei falsi positivi e la conseguente incertezza prognostica causano a breve termine un aumento dell’ansia tra i genitori dei neonati richiamati, mentre non ci sono danni documentati a lungo termine. L'uso proposto di second-tier test di tipo molecolare potrebbe ridurre il tasso di falsi positivi e facilitare la refertazione dei veri positivi (13).
Avendo la potenzialità di identificare in età neonatale anche pazienti LOPD nei quali la sintomatologia clinica si sarebbe manifestata più tardi nella vita, lo SN apre una serie di problematiche irrisolte di natura etica, perché la diagnosi da screening determina un processo di medicalizzazione anticipato, che si accompagna ad un aumento dell’ansia e dello stress genitoriale, nell’attesa del riconoscimento del momento giusto per l’inizio della terapia enzimatica (14).
È auspicabile che in futuro l’identificazione di migliori biomarcatori per la previsione del fenotipo e la personalizzazione del follow-up e trattamento di questi pazienti possano migliorare questi aspetti non risolti (15).
Bibliografia
- van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372(9646):1342-53.
- Hagemans MLC, Winkel LPF, Hop WCJ, et al. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology. 2005; 64(12): 2139–41.
- Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid a-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology. 2007; 68(2): 99–109.
- van der Ploeg AT, Clemens PR, Corzo D, et al. A Randomized Study of Alglucosidase Alfa in Late-Onset Pompe’s Disease. N. Engl. J. Med. 2010; 362(15): 1396–406.
- Chien YH, Hwu WL, Lee NC. Newborn screening: Taiwanese experience. Ann Transl Med. 2019;7(13):281.
- Gragnaniello V, Pijnappel PWWM, Burlina AP, et al. Newborn screening for Pompe disease in Italy: Long-term results and future challenges. Mol Genet Metab Rep. 2022;33:100929.
- Singh S, Ojodu J, Kemper AR, et al. Implementation of Newborn Screening for Conditions in the United States First Recommended during 2010-2018. Int J Neonatal Screen. 2023; 6;9(2):20.
- Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis. 2006;29(2-3):397-404.
- Burton BK, Kronn DF, Hwu WL, Kishnani PS; Pompe Disease Newborn Screening Working Group. The Initial Evaluation of Patients After Positive Newborn Screening: Recommended Algorithms Leading to a Confirmed Diagnosis of Pompe Disease. Pediatrics. 2017;140(Suppl 1):S14-S23.
- Labrousse P, Chien YH, Pomponio RJ, et al. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. MolGenet Metab. 2010;99(4):379-83.
- Niño MY, Wijgerde M, de Faria DOS, et al. Enzymatic diagnosis of Pompe disease: lessons from 28 years of experience. Eur J Hum Genet. 2021;29(3):434-446.
- In 't Groen SLM, de Faria DOS, Iuliano A, et al. Novel GAA Variants and Mosaicism in Pompe Disease Identified by Extended Analyses of Patients with an Incomplete DNA Diagnosis. Mol Ther Methods Clin Dev. 2020;17:337-348.
- Sawada T, Kido J, Nakamura K. Newborn Screening for Pompe Disease. Int J Neonatal Screen. 2020;6(2):31.
- Schoser B, van der Beek NAME, Broomfield A et al. Start, switch and stop (triple-S) criteria for enzyme replacement therapy of late-onset Pompe disease: European Pompe Consortium recommendation update 2024. Eur J Neurol. 2024;31(9):e16383.
- Tarallo A, Carissimo A, Gatto F, et al. microRNAs as biomarkers in Pompe disease. Genet Med. 2019;21(3):591-600.