




la Newsletter

Elena Verrecchia1,2, Barbara Siri3, Roberta Taurisano3
1Centro di Continuità Assistenziale e Fragilità, Fondazione Policlinico Universitario A. Gemelli IRCCS, Roma; 2Dipartimento di Scienze Geriatriche ed Ortopediche, Università Cattolica del Sacro Cuore, Roma; 3Unità Operativa Complessa di Malattie Metaboliche ed Epatologia, Ospedale Pediatrico Bambino Gesù IRCCS, Roma
PKU: gestione del paziente in fase di transizione dall’età...
La transizione è un percorso piuttosto lungo e richiede la...
Descriviamo il caso di un paziente di 22 anni con screening neonatale positivo per fenilchetonuria (Phe 1.314 μmol/L) confermata geneticamente dalla presenza di un’eterozigosi composta in PAH. Seguito presso il Centro dell’Ospedale Pediatrico Bambino Gesù di Roma (OPBG), sin dalla nascita il paziente ha intrapreso una dietoterapia con apporto di Phe pari a circa 500 mg/die integrata con equivalente proteico mostrando buona compliance e valori di Phe costantemente <600 μmol/L.
Non ha inoltre presentato una risposta al test prolungato con BH4. Tuttavia, a partire dall’età di 20 anni ha iniziato a presentare una scarsa compliance al regime dietetico controllato. Pertanto, dopo il riscontro di valori di Phe persistentemente >1.000 μmol/L per 3 mesi consecutivi, nonostante le raccomandazioni ad una corretta gestione della dieta, veniva posta indicazione alla terapia enzimatica sostitutiva con pegvaliase.
In relazione alle indicazioni, alla prescrivibilità del farmaco riservato a pazienti di età superiore a 16 anni, nonché alla gestione dei potenziali eventi avversi, è stato necessario avviare la transizione del paziente presso un Centro per adulti in grado di accogliere pazienti con patologie metaboliche rare, individuato nella Fondazione Policlinico Gemelli (FPG). È stata quindi instaurata una collaborazione tra l’OPBG e la FPG con incontri formativi per il personale medico del Centro per adulti sulla patologia, sull’approccio dietetico-terapeutico e sulle caratteristiche dei pazienti. Dopo tale periodo, si è attuata la transizione mediante un incontro informativo presso l'OPBG per la presentazione del Centro al paziente, ed un incontro di affidamento del paziente al Centro per adulti.
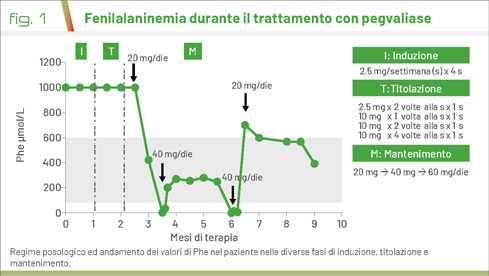 La terapia enzimatica sostitutiva è stata quindi iniziata presso FPG, con aggiornamenti continui tra FPG e OPBG durante la fase di induzione, titolazione e mantenimento (Fig. 1). Soprattutto in quest’ultima fase, i due Centri hanno collaborato nel concordare le strategie terapeutiche, regolando la posologia di pegvaliase e la liberalizzazione del regime alimentare in relazione ai valori di Phe raggiunti, dosati periodicamente presso l'OPBG, e responsabilizzando maggiormente il paziente sul suo stato di salute e sulla sua alimentazione.
La terapia enzimatica sostitutiva è stata quindi iniziata presso FPG, con aggiornamenti continui tra FPG e OPBG durante la fase di induzione, titolazione e mantenimento (Fig. 1). Soprattutto in quest’ultima fase, i due Centri hanno collaborato nel concordare le strategie terapeutiche, regolando la posologia di pegvaliase e la liberalizzazione del regime alimentare in relazione ai valori di Phe raggiunti, dosati periodicamente presso l'OPBG, e responsabilizzando maggiormente il paziente sul suo stato di salute e sulla sua alimentazione.
Ad oggi, il paziente esegue terapia con pegvaliase 20 mg/die con un regime alimentare libero e con valori di Phe costantemente <300 μmol/L.
Il processo di transizione nelle malattie metaboliche ereditarie
Le malattie metaboliche ereditarie (MME) sono un eterogeneo gruppo di patologie genetiche che si manifestano e vengono diagnosticate generalmente in età pediatrica.
Sebbene l’incidenza di ciascuna patologia sia bassa (da 1 su 10.000 a 1 su 1.000.000), l’incidenza cumulativa di tutte le MME è alta, essendo compresa tra 1:800 a 1:2.500 nati vivi (1). Il continuo miglioramento dell’assistenza e l’individuazione di nuove strategie terapeutiche in queste patologie hanno condotto negli ultimi decenni ad un aumento della sopravvivenza di questi pazienti: ciò sta ponendo la necessità di definire un programma di transizione dal Centro pediatrico ad uno per adulti. Si tratta di una vera e propria “sfida” che richiede la formazione di figure specialistiche dell’adulto in collaborazione con i Centri pediatrici metabolici. Il processo di transizione è un percorso piuttosto lungo a cui il paziente dovrebbe auspicabilmente essere preparato quanto prima. Il momento “ideale” per l’inizio di tale processo è determinato dalla maturità psico-emotiva del paziente, dalla sua autonomia e dalla stabilità metabolica della sua condizione.
PKU: gestione terapeutica
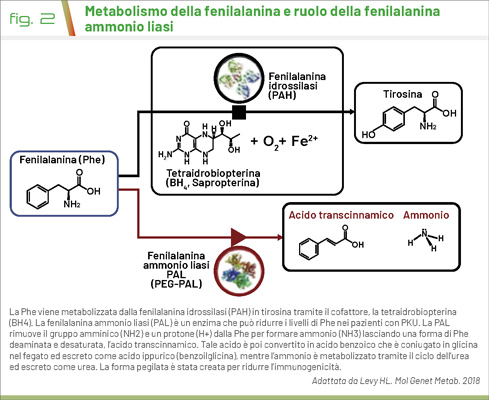
Con una frequenza di circa 1:10.000 nuovi nati, la fenilchetonuria (PKU) rappresenta la prima MME ad essere sottoposta a screening neonatale e diagnosticata in età pediatrica. La PKU è dovuta ad un difetto dell’enzima fenilalanina idrossilasi (PAH) che converte la fenilalanina (Phe) in tirosina, quest’ultima coinvolta nel metabolismo della melatonina e dei neurotrasmettitori cerebrali (2). Questa conversione necessita del cofattore tetraidrobiopterina (BH4). Tale difetto enzimatico porta ad un accumulo di Phe nel sangue e nel sistema nervoso centrale che comporta sintomi quali disabilità intellettiva, epilessia, microcefalia e ipopigmentazione di cute e capelli (3).
 La severità della PKU è storicamente classificata in base ai valori di Phe. La terapia consiste in una dieta ipoproteica a basso contenuto di Phe e di terapia con BH4 in presenza di varianti BH4 responsive (4,5).
La severità della PKU è storicamente classificata in base ai valori di Phe. La terapia consiste in una dieta ipoproteica a basso contenuto di Phe e di terapia con BH4 in presenza di varianti BH4 responsive (4,5).
Negli ultimi anni la gestione terapeutica della PKU ha fatto dei passi avanti con l’approvazione da parte dell’EMA della terapia con pegvaliase, enzima ricombinante di fenilalanina ammonio liasi PEGilata, che converte la Phe in ammoniaca e acido trans-cinnamico (Fig. 2). Tale terapia è stata approvata per i pazienti affetti da PKU con età >16 anni e con valori di Phe persistentemente >600 μmol/L in dietoterapia.
I primi risultati di questa nuova terapia hanno mostrato sicurezza ed efficacia nel controllo dei valori di Phe (6-8). In considerazione della prescrivibilità per pazienti di età superiore a 16 anni, si è reso necessario un processo di transizione dal Centro pediatrico ad uno per adulti.
Conclusioni
La collaborazione costante tra i due Centri OPBG e FPG ratifica il concetto di transizione come un processo continuo di formazione e responsabilizzazione per la gestione delle diverse problematiche in evoluzione, sia per il paziente e la sua famiglia, sia per i medici. Spesso, infatti, nei Centri per l’adulto, seppure esperti nel gestire le complessità e le comorbidità dell'età adulta, manca l’adeguata esperienza nella gestione di alcune problematiche legate alle malattie rare. L’importanza di una assidua collaborazione tra il Centro pediatrico ed il Centro per gli adulti è pertanto fondamentale per garantire una continuità assistenziale di qualità, adattata alle esigenze del paziente adulto (9).
Bibliografia
- Stepien KM, Kieć-Wilk B, Lampe C. Challenges in Transition From Childhood to Adulthood Care in Rare Metabolic Diseases: Results From the First Multi-Center European Survey Front Med (Lausanne). 2021;8:652358
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104(Suppl):S2–9
- de Groot MJ, Hoeksma M, Blau N, et al. Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab. 2010:99 Suppl 1:S86-9.
- van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162.
- Blau N, Belanger-Quintana A, Demirkol M, et al. Management of phenylketonuria in Europe: survey results from 19 countries. Mol Genet Metab. 2010;99(2):109–15.
- Harding CO, Longo N, Northrup H, et al. Pegvaliase for the treatment of phenylketonuria: Final results of a long-term phase 3 clinical trial program. Mol Genet Metab Rep. 2024;39:101084.
- Scala I, Brodosi L, Gueraldi D, et al. Pegvaliase therapy for phenylketonuria: Real-world case series and clinical insights. Mol Genet Metab. 2024;142(1):108151.
- Scala I, Brodosi L, Rovelli V, et al. Management of patients with phenylketonuria (PKU) under enzyme replacement therapy: An Italian model (expert opinion). Mol Genet Metab Rep. 2024;39:101065.
- American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians-American Society of Internal Medicine. A consensus statement on health care transitions for young adults with special health care needs. Pediatrics. 2002;110(6 Pt 2):1304-6.