




la Newsletter

Andrea Biuso1,2, Elisabetta Prada2
1Ospedale dei Bambini Buzzi, Dipartimento di Scienze Biomediche e Cliniche, Università degli Studi di Milano; 2UOC Pediatria – Centro Fondazione Mariani per il Bambino Fragile, ASST-Lariana, Como
Quando l’occhio fa da “spia”: emeralopia e deficit...
La sindrome di Bardet-Biedl ha natura pleiotropica e richiede...
L. è un primogenito di genitori sani non consanguinei nato a termine da parto eutocico dopo gravidanza normodecorsa, parametri alla nascita e decorso perinatale nella norma. Riscontro alla nascita di esadattilia post-assiale del piede sinistro corretta chirurgicamente. È seguito dall’età di 6 anni da un nutrizionista per sovrappeso con tendenza all’iperfagia e preso in carico da un punto di vista logopedico per lievi difficoltà fonetiche. All’età di 8 anni ha iniziato a lamentare difficoltà a leggere la lavagna e disturbo della visione durante le ore notturne.
La valutazione oculistica ha messo in luce miopia; il fundus oculi e la camera anteriore sono risultati nella norma. Per ulteriore peggioramento dell’acuità visiva ai campi periferici eseguiti approfondimenti con tomografia ottica computerizzata (OCT) ed elettroretinogramma (ERG), che hanno mostrato reperti compatibili con quadro di distrofia retinica.
A 10 anni giunge presso il nostro centro per inquadramento diagnostico. Alla valutazione clinica: peso 90-97°p.le, altezza 75°p.le, BMI >97°p.le. Al volto non presenti franchi dismorfismi, al cuore soffio sistolico 2/VI, micropene con testicoli in sede, assenza di segni di sviluppo puberale, restante obiettività clinica nei limiti di norma. A completamento, eseguiti esami ematochimici comprendenti profilo gluco-lipidico (modesta ipercolesterolemia) e studio dell’asse gonadotropine-testosterone (valori compatibili con età pre-pubere); ecografia addome con studio dell’apparato urinario e visita otorino-laringoiatrica con test audiometrico, risultati nella norma.
In considerazione del sospetto clinico eseguito pannello NGS multigenico che ha confermato una variante omozigote di significato verosimilmente patogenetico a carico del gene BBS1.
La sindrome di Bardet-Biedl: epidemiologia e genetica
La sindrome di Bardet-Biedl (BBS) è un raro disordine ereditario a coinvolgimento multisistemico appartenente alle ciliopatie primarie, con trasmissione autosomica recessiva ed espressività variabile (1). La prevalenza è stimata tra 1:120.000 e 1:160.000 e tende ad aumentare in comunità con alta percentuale di unioni tra consanguinei (2). Attualmente sono stati identificati almeno 28 geni implicati nella patogenesi della BBS, che per la maggior parte codificano per proteine indispensabili alla costituzione di una struttura ottamerica chiamata BBSoma, cruciale per la funzionalità delle ciglia primarie (3).
Presentazione clinica e diagnosi
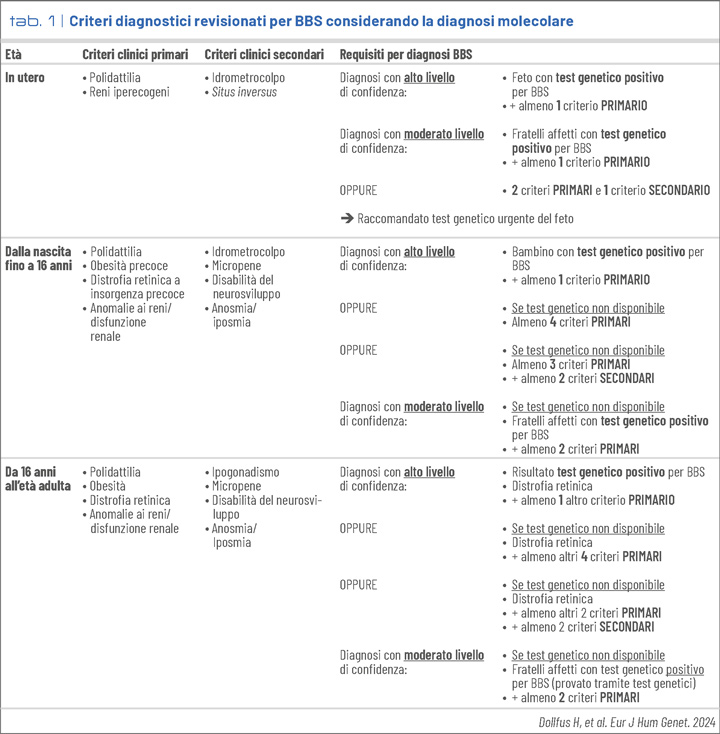
La tabella 1 mostra le principali caratteristiche cliniche identificate per età ed i criteri diagnostici per la BBS recentemente elaborati in una Clinical Consensus Statement (1).
Diagnosi differenziale
Le condizioni in diagnosi differenziale con la BBS sono: la sindrome di Alström (per retinopatia, obesità, ipoacusia), la sindrome di McKusick-Kaufman e la sindrome di Meckel (per polidattilia ed anomalie renali), la sindrome di Joubert (per retinopatia, polidattilia ed anomalie renali), l’amaurosi congenita di Leber (per retinopatia) (2-5).
Assistenza e terapia
La BBS ha una natura pleiotropica e richiede, sin dalle prime manifestazioni, una gestione multidisciplinare (oculistica, nutrizionale, nefrologica, endocrinologica, neuropsichiatrica) con un follow-up personalizzato (2).
Attualmente non è disponibile una terapia specifica per questa condizione. Per la degenerazione retinica risulta di fondamentale importanza impostare un programma educativo e di supporto precoce per ridurre l’impatto della perdita visiva (p.es. sistema Braille, strumenti informatici).
Il futuro è rappresentato dalla ricerca in ambito di terapia genica, alla luce dei promettenti risultati ottenuti per il trattamento di altre forme di distrofia retinica ereditaria (2,4).
Segnaliamo la presenza dell'Associazione Sindrome Bardet-Biedl Italia APS (A.S.B.B.I. Aps) attiva sul territorio nazionale dal 2009 e impegnata nell'attività di prima accoglienza per le famiglie delle persone affette da tale sindrome, promuovendo occasioni di scambio e confronto tra personale sanitario e genitori.
Per contattare l'Associazione: www.asbbi.it - info@asbbi.it
Bibliografia
- Dollfus H, Lilien MR, Maffei P, et al. Bardet-Biedl syndrome improved diagnosis criteria and management: Inter European Reference Networks consensus statement and recommendations. Eur J Hum Genet. 2024;32(11):1347-1360.
- Melluso A, Secondulfo F, Capolongo G, et al. Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook. Ther Clin Risk Manag. 2023;19:115-132.
- Tomlinson JW. Bardet-Biedl syndrome: A focus on genetics, mechanisms and metabolic dysfunction. Diabetes Obes Metab. 2024;26 Suppl 2:13-24.
- Forsyth R, Gunay-Aygun M. Bardet-Biedl Syndrome Overview. 2003 Jul 14 [updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024.
- Forsythe E, Sparks K, Best S, et al. Risk Factors for Severe Renal Disease in Bardet-Biedl Syndrome. J Am Soc Nephrol. 2017;28(3):963-970.