




la Newsletter

Paolo Fontana1, Fortunato Lonardo1, Francesca Del Vecchio Blanco2, Margherita Russo2
1Genetica Medica - A.O.R.N. San Pio, Benevento; 2Genetica Medica - AOU Policlinico, Università degli Studi della Campania "L. Vanvitelli"
Sindrome CODAS: una nuova entità nosologica da considerare in...
L’acronimo CODAS sta per “Cerebral, Ocular, Dental, Auricular,...
Giungeva a visita genetica una bambina di 33 mesi, nata da una seconda gravidanza di genitori non consanguinei; la prima gravidanza era terminata con un aborto spontaneo. La bambina era nata da taglio cesareo d’elezione a termine di una gravidanza normodecorsa. Il peso alla nascita era di 3,720 Kg (+0,7 DS); la lunghezza 51 cm (+0,53 DS); la circonferenza cranica 35,5 cm (+1,31 DS). All'età di tre mesi la paziente aveva ricevuto diagnosi di cataratta congenita bilaterale, trattata chirurgicamente. Agli ultimi controlli si segnalano anche strabismo bilaterale e nistagmo.
Intorno all’anno di età era stato intrapreso un follow-up neuropsichiatrico per ritardo dello sviluppo psicomotorio ed ipotonia assiale. Una risonanza magnetica encefalica evidenziava una atrofia cerebellare, con interessamento sia del verme che degli emisferi. Gli EEG segnalano invece anomalie epilettiformi sui due emisferi, sebbene fino ad ora la bambina non abbia manifestato episodi critici. Tra i vari controlli si segnalano anche potenziali evocati uditivi (Auditory Brainstem Response, ABR), ecografia cardiaca ed ecografia addominale, tutti nella norma. Nel corso del tempo il ritardo psicomotorio si è reso più evidente: il linguaggio è povero, ma presente; persiste l’ipotonia, seppur più sfumata in seguito alle terapie; la deambulazione è difficoltosa, anche per via della ridotta capacità visiva. Attualmente la bambina pratica fisiokinesiterapia, psicomotricità, logopedia.
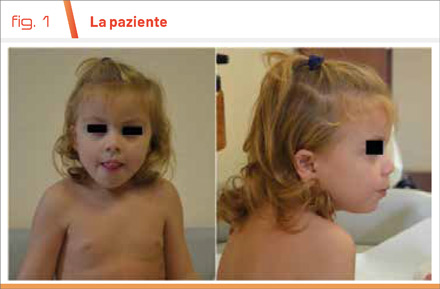 Al nostro esame clinico (Fig. 1) rilevavamo una lunghezza di 91 cm (-0,5 DS), un peso di 13,1 Kg (-0,1 DS), una circonferenza cranica di 48 cm (-0,2 DS), atteggiamento scoliotico, ipotonia, andatura atassica, piede talo-valgo bilaterale, linguaggio molto povero e limitato ad alcune paroline, carattere socievole. Si procedeva in prima battuta con uno SNP-array, da cui non emergevano riarrangiamenti patogenetici. In seguito veniva eseguito un esoma clinico, che evidenziava uno stato di eterozigosi composta del gene LONP1: c.1752C>A (esone 11), p.Asn584Lys (paterna) e c.2345C>T (esone16), p.Thr782Ile (materna).
Al nostro esame clinico (Fig. 1) rilevavamo una lunghezza di 91 cm (-0,5 DS), un peso di 13,1 Kg (-0,1 DS), una circonferenza cranica di 48 cm (-0,2 DS), atteggiamento scoliotico, ipotonia, andatura atassica, piede talo-valgo bilaterale, linguaggio molto povero e limitato ad alcune paroline, carattere socievole. Si procedeva in prima battuta con uno SNP-array, da cui non emergevano riarrangiamenti patogenetici. In seguito veniva eseguito un esoma clinico, che evidenziava uno stato di eterozigosi composta del gene LONP1: c.1752C>A (esone 11), p.Asn584Lys (paterna) e c.2345C>T (esone16), p.Thr782Ile (materna).
La presenza di due varianti del gene LONP, ha consentito di confermare la diagnosi di una condizione sindromica molto rara nota come sindrome CODAS (MIM 600373).
La sindrome CODAS
L’acronimo CODAS sta per “Cerebral, Ocular, Dental, Auricular, Skeletal anomalies” ed è stato coniato per indicare una rara patologia monogenica, autosomica recessiva, associata all’interessamento dei suddetti organi e apparati, e secondaria ad una disfunzione mitocondriale dovuta a varianti con perdita di funzione del gene LONP1.
Il primo lavoro scientifico in cui viene utilizzata l’espressione “CODAS syndrome” risale al 1991, quando Shebib e colleghi descrissero un bambino con ritardo psicomotorio, cataratta bilaterale, ptosi palpebrale, solco mediano nasale, orecchie malformate, ipoacusia neurosensoriale, anomalie dei denti, bassa statura, anomalie di ossificazione vertebrale, displasia dell’anca. Negli anni successivi le ulteriori segnalazioni di questa condizione sono state pochissime, a conferma della rarità del disordine.
Prima che venisse identificato il gene malattia la diagnosi era essenzialmente clinica e venivano pertanto diagnosticati come CODAS soltanto i pazienti con una facies molto peculiare e tipica di questa condizione.
Decorso clinico
Alla nascita i bambini presentano in genere delle orecchie a basso impianto, accartocciate e con elice ripiegato, un ponte nasale piatto con un solco verticale mediano, ptosi palpebrale, epicanto, in alcuni casi microcefalia. Tra i 2 e i 6 mesi di vita si sviluppa rapidamente una cataratta nucleare bilaterale. I denti erompono in ritardo, hanno spesso una conformazione anomala, con protrusioni dello smalto a partire dalla punta delle cuspidi.
Nel primo anno di vita emerge un ritardo psicomotorio, spesso moderato o grave, che può associarsi ad ipotonia e, talvolta, ad epilessia. Le anomalie scheletriche comprendono bassa statura, displasia metafisaria ed epifisaria, scoliosi, ginocchio valgo. In circa la metà dei casi è stata riportata una ipoacusia neurosensoriale. Nella sua forma classica sono rare le complicanze gravi, tra cui va segnalato un aumentato rischio di ostruzione laringea nei primi giorni di vita. Vi sono però alcune forme atipiche con prognosi meno favorevole.
Il gene LONP1
Nel 2015 è stato identificato il gene-malattia della sindrome CODAS. Il gene LONP1 (19p13.3, MIM 605490) codifica per la proteasi mitocondriale ATP-dipendente Lon, la quale esercita una funzione proteolitica ed agisce prevalentemente nei mitocondri, dove contribuisce allo smaltimento delle proteine danneggiate, ossidate o mal ripiegate, e alla preservazione del corretto numero di copie di DNA mitocondriale. La proteasi Lon possiede anche una attività di proteina chaperone e favorisce l’assemblaggio dei complessi della catena respiratoria mitocondriale.
La perdita di funzione del gene LONP1 determina, a livello cellulare, una ridotta quantità di DNA mitocondriale, un malfunzionamento della catena respiratoria mitocondriale, un difetto di produzione di ATP e del trasporto mitocondriale. Il gene è espresso anche a livello del nucleo dove partecipa alle risposte cellulari allo stress ossidativo indotto da radicali liberi dell’ossigeno (ROS).
Diagnosi differenziale
Il quadro clinico è molto peculiare e differisce quasi sempre dalla tipica presentazione delle malattie mitocondriali, che per solito si associano a grave acidosi lattica e coinvolgimento muscolare, neurologico, epatico, oculare. Nella sindrome CODAS invece le caratteristiche faciali, dentali e scheletriche sono considerate patognomoniche.
Spesso nei primi mesi di vita la cataratta è l’unico segno clinico, per cui la condizione può andare in diagnosi differenziale con le forme di cataratta congenita isolata.
Quando le caratteristiche faciali sono invece più marcate la diagnosi differenziale è con altre sindromi genetiche come la Kabuki, per il distretto oculare, o la CHARGE, per l’aspetto delle orecchie. Quando il coinvolgimento scheletrico è precoce può insorgere il sospetto di alcune displasie ossee tra cui la condrodisplasia puntata.
Forme atipiche
In seguito alla identificazione del gene-malattia e all’avvento del sequenziamento NGS in diagnostica, sono state identificate varianti patogenetiche del gene LONP1 anche in pazienti che, dal punto di vista clinico, non avrebbero indotto al sospetto di sindrome CODAS. È evidente che varianti differenti in siti differenti della proteina determinano una notevole variabilità del fenotipo. Sono stati descritti, ad esempio, pazienti con sintomatologia più sfumata, interessamento neurologico lieve e assenza delle tipiche caratteristiche ossee e dentarie. In altri pazienti è stato osservato un coinvolgimento esclusivamente neurologico, con disabilità intellettiva severa, ipotonia e atrofia cerebellare progressiva. Alcune specifiche varianti del gene LONP1 sono state invece associate a cataratta congenita isolata. Infine, ci sono state alcune recenti segnalazioni di pazienti CODAS con quadro clinico molto più simile ad altre malattie mitocondriali, con acidosi lattica congenita, ipotonia profonda, debolezza muscolare, disabilità intellettiva grave, atassia.
Diagnosi, trattamento e rischio di ricorrenza
La diagnosi è esclusivamente molecolare, con analisi specifica del gene LONP1 nei casi familiari o quando il quadro clinico è molto eclatante, oppure, nella maggior parte dei casi, mediante sequenziamento dell’esoma o del genoma. Il trattamento è sintomatico e prevede un approccio multidisciplinare. È richiesto il follow-up da parte del neuropsichiatra infantile, dell’oculista, dell’odontoiatra, dell’audiologo, dell’ortopedico.
Trattandosi di una patologia recessiva, tutti i pazienti con sindrome CODAS sono omozigoti oppure eterozigoti composti per varianti patogenetiche del gene LONP1. In genere i soggetti affetti hanno due genitori entrambi eterozigoti per una variante nel gene LONP1. In queste coppie il rischio di ricorrenza per gravidanze successive è del 25%.
Bibliografia
- Dikoglu E, Alfaiz A, Gorna M, et al. Mutations in LONP1, a mitochondrial matrix protease, cause CODAS syndrome. Am J Med Genet A. 2015;167(7):1501-9.
- Strauss KA, Jinks RN, Puffenberger EG, et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Hum Genet. 2015;96(1):121-35.
- Inui T, Anzai M, Takezawa Y, et al. A novel mutation in the proteolytic domain of LONP1 causes atypical CODAS syndrome. J Hum Genet. 2017;62(6):653-655.
- Khan AO, AlBakri A. Clinical features of LONP1-related infantile cataract. J AAPOS. 2018;22(3):229-231.
- Peter B, Waddington CL, Oláhová M, et al. Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum Mol Genet. 2018;27(10):1743-1753.