




la Newsletter

Irene Ranzetti1, Alessandra Grassi2, Elisabetta Prada3
1Università degli Studi di Trieste, Specializzanda in Pediatria;
2Università degli Studi dell’Insubria, Specializzanda in Pediatria;
3UOC Pediatria – Centro Fondazione Mariani per il Bambino Fragile, ASST-Lariana, Como
Sindrome di Koolen-de Vries | La gestalt indirizza il sospetto...
La gestalt indirizza il sospetto diagnostico, ma la conferma...
G. è una secondogenita nata da gravidanza gemellare monocoriale monoamniotica, da genitori sani non consanguinei. Nata da taglio cesareo all’età gestazionale di 33 settimane, peso alla nascita 1.860 g, APGAR 9/9. Alimentata via sondino naso gastrico (SNG) e ricoverata in Terapia intensiva neonatale (TIN) per 3 settimane; a 2 mesi posta diagnosi di laringomalacia e reflusso gastroesofageo (RGE). A 3 anni comparsa di crisi tonico-cloniche, responsive a valproato.
G. condivide con la gemella l’ipotono e il ritardo psicomotorio, più accentuato nel linguaggio.
A 10 anni, quando giungono al nostro Centro per la presa in carico, la diagnosi è chiara: la loro storia, il naso piriforme, le fessure palpebrali strette e l’astigmatismo ci convincono sia sindrome di Koolen-de Vries.
La sindrome
La sindrome di Koolen-de Vries (KdVS), anche nota come microdelezione 17q21.31, è una condizione genetica rara multisistemica, caratterizzata da ritardo psicomotorio/disabilità intellettiva, ipotonia, caratteristiche facciali tipiche. Il quadro può associarsi a malformazioni congenite, epilessia, disturbi del visus. Il primo caso fu descritto nel 2006, ad oggi ne sono noti oltre 200.
Elementi di sospetto
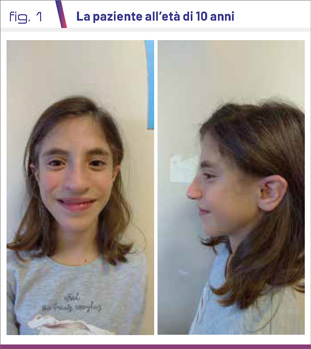 La KdVS va sospettata in presenza di disabilità intellettiva lieve-moderata, dismorfismi e ipotono nella prima infanzia con difficoltà di alimentazione. Le note dismorfiche distintive sono il volto allungato con rime palpebrali strette e upslanting, epicanto, ptosi, naso piriforme con punta bulbosa e radice nasale ampia e alta, columella lunga, orecchie grandi e prominenti (Fig. 1). Le dita sono tipicamente lunghe e affusolate. Il sospetto clinico è rafforzato dalla presenza di anomalie muscolo-scheletriche, cardiache, genito-urinarie, alterazioni del visus o epilessia.
La KdVS va sospettata in presenza di disabilità intellettiva lieve-moderata, dismorfismi e ipotono nella prima infanzia con difficoltà di alimentazione. Le note dismorfiche distintive sono il volto allungato con rime palpebrali strette e upslanting, epicanto, ptosi, naso piriforme con punta bulbosa e radice nasale ampia e alta, columella lunga, orecchie grandi e prominenti (Fig. 1). Le dita sono tipicamente lunghe e affusolate. Il sospetto clinico è rafforzato dalla presenza di anomalie muscolo-scheletriche, cardiache, genito-urinarie, alterazioni del visus o epilessia.
Criteri diagnostici
Attualmente non vi è un consensus sui criteri diagnostici della KdVS. La gestalt indirizza il sospetto, ma la conferma deriva dal riscontro genetico di delezione cromosomica/mutazione a carico del gene KANSL1 che ne determina l’aploinsufficienza.
Auxologia
Pur essendo spesso scarsa nella prima infanzia per le difficoltà di alimentazione, la crescita tende a regolarizzarsi nelle epoche successive ed è raro il riscontro di bassa statura nell’adulto.
Malformazioni maggiori
Non esiste una malformazione patognomonica.
Circa il 40% dei pazienti mostra un coinvolgimento cardiaco, prevalentemente difetto inter-atriale (DIA)/ difetto inter-ventricolare (DIV).
Il criptorchidismo è la malformazione genito-urinaria più comune (50% dei maschi affetti); seguono reflusso vescico ureterale, ipospadia, idronefrosi e, meno frequentemente, doppio ristretto renale, pielectasia, macrorchidismo. Rari i casi di nefropatia da reflusso associati ad ipertensione. Rare malformazioni del SNC (ventricolomegalia, ipo/aplasia del corpo calloso, malformazioni tipo Arnold-Chiari 1, idrocefalo, emorragie intraventricolari).
Complicanze mediche associate
Epilessia e disturbi del visus interessano circa metà della popolazione. Le crisi, generalizzate o cloniche unilaterali, sono di norma responsive ai farmaci e vanno indagate mediante EEG in sonno-veglia. I casi neurologicamente più compromessi possono sottendere malformazioni del SNC.
I difetti visivi più comuni sono miopia e strabismo; rari i casi di ipovisus da causa malformativa (cataratta congenita, atrofia ottica) o danno centrale. Talvolta è presente ipoacusia, di solito trasmissiva.
Circa l’80% dei pazienti con KdVS presenta problematiche muscolo-scheleteriche di entità variabile a carico di rachide (scoliosi/cifosi), torace (pectus excavatum/carinatum), piedi (piattismo, piede cavo/torto) ed articolazioni (lussazione d’anca, iperlassità legamentosa). Segnalati casi di tracheo/laringomalacia.
Sviluppo psicomotorio
È presente ritardo psicomotorio che evolve in disabilità intellettiva lieve-moderata con maggiore compromissione dell’area del linguaggio. Tipica nella prima infanzia, l’ipotonia (> 80%) causa suzione ipovalida. Pur migliorando con la crescita, l’ipotonia può ancora condizionare scialorrea, difficoltà nella gestione dei pasti (masticazione, deglutizione) e delle secrezioni respiratorie con ricorrenza delle infezioni. I bambini presentano spesso un temperamento socievole e affettuoso. In adolescenza si segnalano problematiche neuropsicologiche [ansia, disturbo da deficit di attenzione/iperattività (ADHD), comportamenti stereotipati] ed evoluzione talvolta in età adulta in senso psichiatrico o regressivo.
Difetto genetico
La sindrome è causata dall’aploinsufficienza del gene KANSL1, localizzato sul cromosoma 17 nella regione q21.31, determinata da una delezione dell’intera regione cromosomica (80% dei casi) o da una mutazione loss-of-function a carico del gene, tipicamente de novo. Benché la del17q21.31 provochi la perdita di altri quattro geni, di cui due (CRHR1, MAPT) coinvolti in processi di neurodegenerazione, KANSL1 sembra essere il main gene responsabile.
La conferma diagnostica avviene mediante sequenziamento diretto del gene KANSL1 (in caso di forte sospetto clinico) o mediante array-CGH.
Sostegno
Dal 2016 esiste l’associazione Kool Kids KANSL1 Italia (www.koolkidsitalia.org), nata per offrire sostegno, informazione e condivisione di esperienze alle famiglie di bambini affetti da KdVS.
Bibliografia
- Koolen DA, Pfundt R, Linda 1K, et al. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet. 2016;24(5):652-9.
- Koolen DA, Morgan A, de Vries BBA. Koolen-de Vries Syndrome. 2010 Jan 26 [updated 2023 Feb 2]. In: Adam MP, Feldman J, Mirzaa GM, et al. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. PMID: 20301783.
- Sharp AJ, Hansen S, Selzer RR, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38(9):1038-42.